ЛОГИЧЕСКАЯ И ОНТОЛОГИЧЕСКАЯ СТРУКТУРА ДЛЯ ИССЛЕДОВАНИЯ ГЕНЕТИЧЕСКИХ РЕГУЛЯТОРНЫХ СЕТЕЙ НА ПРИМЕРЕ СИСТЕМЫ ЖЕЛЧНЫХ КИСЛОТ И КСЕНОБИОТИКОВ (BAXS)

DOI: https:/doi.org/10.18454/jbg.2020.2.14.1
López J.1 *, Ramírez Y.2, Dávila J.3, Bastidas M.4
1, 2 LCAR: High Performance Computing Laboratory, National Experimental University of Táchira, San Cristóbal, Táchira, Venezuela;
3 CESIMO: Center for Simulation and Modeling, University of Los Andes, Mérida, Venezuela;
4 LaBioMex: Laboratory of Biology and Experimental Medicine. University of Los Andes, Mérida, Venezuela
* Correspodning author (jlopez@unet.edu.ve)
Received: 20.11.2020; Accepted: 30.11.2020; Published: 14.12.2020
A LOGICAL AND ONTOLOGICAL FRAMEWORK FOR KNOWLEDGE DISCOVERY ON GENE REGULATORY NETWORKS.
CASE STUDY: BILE ACID AND XENOBIOTIC SYSTEM (BAXS)
Research article
Abstract
This work aims to develop a novel computational framework for automatic or semi-automatic modeling of gene regulatory networks, in which possible connections between genetic structural knowledge and other forms of knowledge available about GRNs can be explored. To perform the modeling of such networks, we consult computing services available through portals on the Internet. Some of these portals are: GeneOntology, PDB, HGNC, PathwayCommons, UniProt, PubMed, Mesh, among others. The mentioned sites provide services that make possible automatic access and can, therefore, be used to organize knowledge bases that integrate their resources. Our team has developed a prototype system that allows such an integration and the analysis of information so obtained in different modalities. In our case, the knowledge automatically gathered and modeled, expresses the identity of the objects on a network and their interactions, as well as their biological functions, biological processes and cellular components. In this sense, our proposal aims at the semantic modeling of GRNs by layers, each layer representing a standard level of description. It starts with a basic level for the model of the transcription regulatory region and goes up to a level for extra-cellular objects. Our platform consolidates all the information obtained from the different sources mentioned into one integrated representation. This representation can then be used for the discovery of biological signaling pathways and the discovery of regulatory sub-networks. At the moment, we are running processes to model and analyze the Bile Acid and Xenobiotic System (BAXS), and some results are already available. For instance, given a set of ligands, a couple of proteins and their related DNA sequences, it is possible: a) to discover a GRN describing how the given proteins regulate each other; and b) to produce sets of user-validated pathways for the proteins under consideration. Details about our system, named biopatternsg (biopatterns searching), can be viewed at: https://github.com/biopatternsg/biopatternsg.
Keywords: gene regulatory network, ontology, modeling, inference, information retrieval.
Лопес Х.1 *, Рамирес Й.2, Давила Х.3, Бастидас М.4
1, 2 LCAR: Высокопроизводительная вычислительная лаборатория, Национальный экспериментальный университет Тачиры, Сан-Кристобаль, Тачира, Венесуэла;
3 CESIMO: Центр моделирования, Андский университет, Мерида, Венесуэла;
4 LaBioMex: Лаборатория биологии и экспериментальной медицины. Андский университет, Мерида, Венесуэла
* Корреспондирующий автора (jlopez@unet.edu.ve)
Получена: 20.11.2020; Доработана: 30.11.2020; Опубликована: 14.12.2020
ЛОГИЧЕСКАЯ И ОНТОЛОГИЧЕСКАЯ СТРУКТУРА ДЛЯ ИССЛЕДОВАНИЯ ГЕНЕТИЧЕСКИХ РЕГУЛЯТОРНЫХ СЕТЕЙ
НА ПРИМЕРЕ СИСТЕМЫ ЖЕЛЧНЫХ КИСЛОТ И КСЕНОБИОТИКОВ (BAXS)
Научная статья
Аннотация
Данная работа направлена на разработку новой вычислительной структуры для автоматического или полуавтоматического моделирования генетических регуляторных сетей, в которых могут быть выявлены всевозможные виды информации как о генетических структурах, так касательно генетических регуляторных сетей. Для моделирования данных сетей были использованы специализированные интернет-ресурсы. В список использованных интернет-ресурсов входят GeneOntology, PDB, HGNC, PathwayCommons, UniProt, PubMed, Mesh и др. Данные сайты предоставляют свободный доступ к данным и, следовательно, могут быть использованы для организации баз знаний, интегрирующих их ресурсы. Команда специалистов данного исследования разработала прототип системы, которая позволяет осуществлять такую интеграцию и анализировать полученную таким образом информацию в различных модальностях. В случае данного исследования собираемая и моделируемая информация выражает идентичность объектов и их взаимодействие в генетической сети, а также их биологические функции, процессы и клеточные компоненты. Таким образом, проводимое исследование направлено на семантическое моделирование генетических регуляторных сетей методом информационных слоёв, каждый из которых представляет собой стандартный уровень описания. Моделирование начинается с базового уровня модели регуляции транскрипции и поднимается до уровня внеклеточных объектов. Разработанная платформа объединяет всю полученную из упомянутых источников информацию в одну интегрированную модель. Данная модель может быть использована для нахождения биологических путей передачи сигнала и регуляторных подсетей. В настоящее время активным является процесс моделирования и анализа желчных кислот и ксенобиотической системы (BAXS). Некоторые результаты уже доступны. Например, имея набор лигандов, несколько белков и связанные с ними последовательности ДНК, можно: а) найти генетическую регуляторную сеть, описывающую, каким образом данные белки взаимодействуют друг с другом; б) создать пользовательские наборы путей для рассматриваемых белков. Подробную информацию о разработанной системе под названием biopatternsg (поиск биопаттернов) можно получить, пройдя по ссылке: https://github.com/biopatternsg/biopatternsg.
Ключевые слова: генная регуляторная сеть, онтология, моделирование, вывод, поиск информации.
1. Introduction
The computational and biological advances of the last decades have allowed the modeling of the interaction maps of molecular systems of many living beings. Examples of this are to be found in the Pathway Commons and the Reactome Projects [1], [2]. Some of the projects involved specific knowledge representation strategies such as the semantic web and ontologies [3], [4], [5]; Natural Language Processing (NLP) [6], [7], [8], [9], [10], [11, [12], [13]; and, of particular interest here, logical-based modeling and analyzing systems [14], [15], [16], [17], [18], [19], [20]. There clearly is a trend for the development of tools for the discovering of relations between biological objects and the productions of molecular interaction maps, including modeling behaviors in the presence of external agents, as in the case of drugs [2], [21], [22], [23], [24].
Gene Regulatory Networks (GRNs) are a special case of Molecular Interaction Map (MIM) [25]. In such networks a particular product (a transcript) relates to the regulation of other objects in the same network or in different networks. Therefore, a regulation event can activate a product that in turn participates in another event, which activates or inhibits another product. It is normal then that in such networks the complexity of the interrelationships grows very quickly when modeling. In order to manage the current knowledge inherent in GRNs, computer strategies have been developed aimed at the description, organization, interrelation and analysis of the elements that constitute them. Among these strategies are the ontologies [4], [26], and the process diagrams [21], [22], [23]; the first oriented to the semantic analysis of the maps and the second to the simulation of their corresponding biochemical dynamics.
We build upon those tendencies by aiming at the automatic construction of knowledge bases, modeling networks of molecular interactions as described by the scientific literature. In short, we have developed a software pipeline that reads regular, scientific abstracts from papers and produces computationally verifiable representations of GRNs.
Generally a GRN is built for particular biological subsystems, responsible for the regulation of processes such as apoptosis, bile acid production, the generation of hormones or antibodies, among others. In each case a GRN must be modeled and analyzed while integrating different sources of information [25]. This task may involve using tools such as Prótége [21], [22], [24], [27] and web services like those offered by portals like PubMed [28]; the Protein Data Bank (PDB) [29], [30]; HGNC (HUGO Gene Nomenclature Committee) [31]; GeneOntology [32]; Uniprot [33]; Mesh [34] and Pathway Commons [1].
Thus, there exists different approaches and tools for the automatic modeling of GRNs, still functionally separated from the information contained in scientific publications, which must be read and assimilated by humans before their information can be (re)integrated into experimental processes. In such cases, data and expert knowledge is put together by humans who perform a sort of reverse engineering of a biological process, which is then presented as a GRN [6], [7], [8], [10], [11], [12], [19], [25], [35], [36], [37], [38], [39], [40], [41], [42], [43].
Meanwhile portals, such as PubMed, have millions of scientific references in the medical and biological areas describing details of the types of interactions corresponding to specific biological objects.
The goals of the work here presented are: 1) the automatic construction of knowledge bases that describe networks of molecular interactions (molecular species and their possible interactions) and 2) the development of exploratory analysis strategies, which could assist new findings or new conclusions from what is already known (and has been published). As for 1) the developed system collects, from publishing stores and repositories over the Internet, knowledge that may be helpful to answer queries with respect to a specific GRN. For 2) we have designed a set of logical representations of a GRN that can be used to reason about, generalize or explain solutions.
Our purpose is to answer different queries of the style: given a ligand, what type of protein-protein interactions result when that ligand binds to a known receptor, and which one leads to an activation (or inhibition) of the transcriptional response of some gene. And also questions like: is there a subnetwork in the GRN describing interconnected pathways up-regulating (first) and down-regulating (later) a given pair of proteins? This work shows a way to deal with these questions and others. At the moment, the tools presented here only consider deduction as the way to answer the kind of queries mentioned.
Using the schematic strategy expressed in Figure 1, we produce the semantic modeling of a network and we do so by progressively involving certain conceptual levels defined beforehand. One can start from the transcription regulatory DNA sequence of a protein and latter move to levels that involve extra-cellular objects (a drug, for example). As Figure 1 suggests, our methodology starts from a region of the protein's sequence where regulation is likely to occur, defining the transcription factors that recognize it and, from there, the processing moves upwards, by levels, integrating the biological objects that an expert group has provided. Figure 1 also shows that other new objects are added to the network from consulted services like those provided by PDB. A functional prototype of a system that performs all of the above is described in what follows.
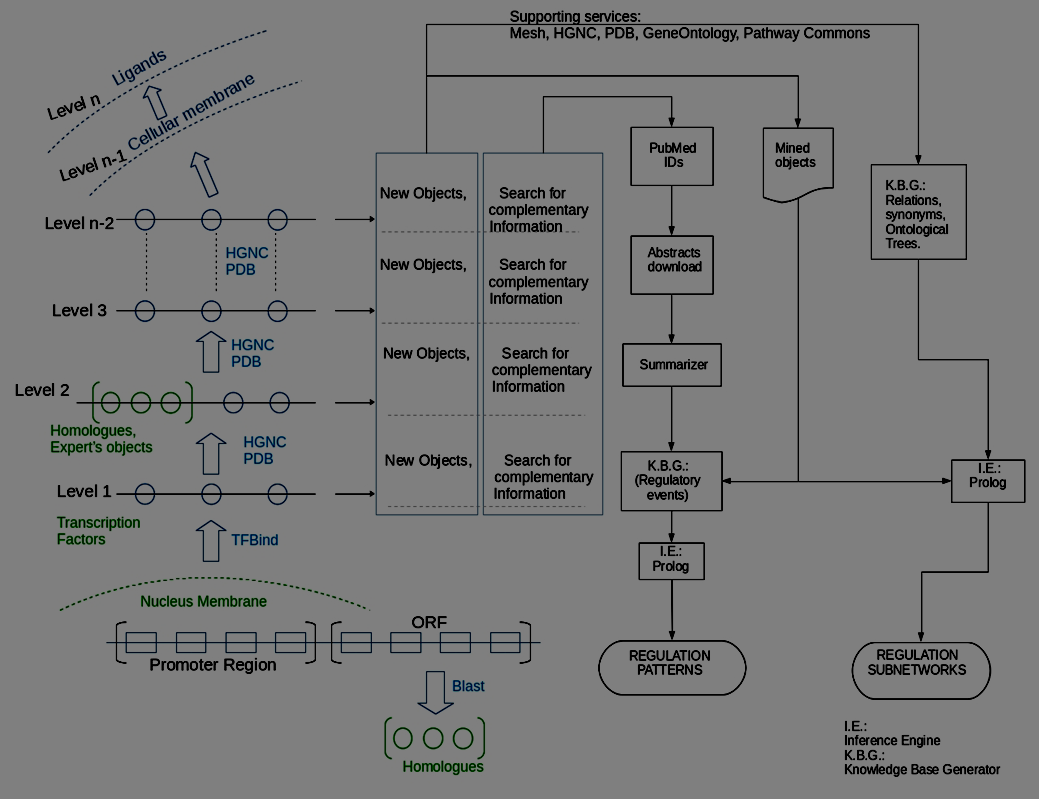
Figure 1 – System architecture and work-flow for the semantic modeling and analyzing of a GRN
1.1. A pipeline of processes
We have implemented a pipeline of automatic processes, i.e. a set of programs that work in tandem to produce and analyze a computationally represented GRN (see Figure 2). The pipeline relies on web services such as PubMed, Mesh, GeneOntology, PDB, Pathway Commons and Uniprot. The pipeline includes a program previously used as a text summarizer [44], [45] to retrieve information from papers written by human experts. The program has been adapted to extract relevant information from the texts. It is a form of Natural Language Processing (NLP) to obtain reports of events of the form event (subject, relationship, object). The above has led to the first implementation of a methodology capable of exploring possible regulatory pathways and the possibility of discovering regulatory subnets within a GRN. The modeling experiment developed here is expected to illustrate the feasibility of the type of automatic modeling and analysis that we propose. The Bile Acid and Xenobiotic System [46], which has already been modeled as a GRN [47], will be used for the purposes of comparison and evaluation of the automatic output.
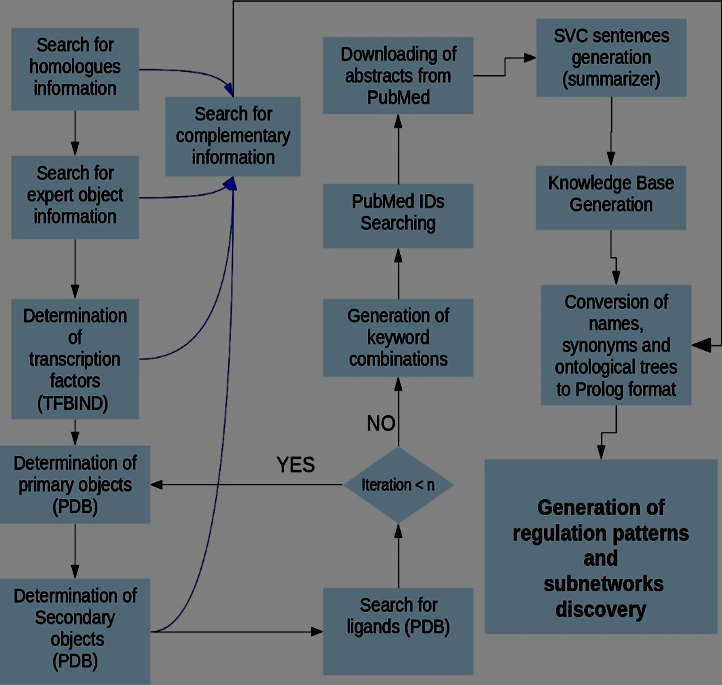
Figure 2 – Pipeline of process proposed to achieve the semantic modeling of a GRN for a targeted protein
1.2. The BAXS system
The Bile Acid and Xenobiotic System (BAXS) (see Figure 3) offers a comparative framework, as we just said. The network described in [47] was modeled by human experts by hand using an ontology designed with the BIOPAX ontology standards [26], [47], a series of queries were implemented using the SPARQL query language [48] and the Pellet inference engine [3]. A subset of those queries are also coded in the present work and some of the results provided by the inference engine we use will be discussed. The approach proposed in this project is different to that exposed in [47] but its results are comparable and extended as new queries and facilities are included. In the next lines we present some comments about how the system automatically organizes and uses the knowledge collected for the BAXS system.
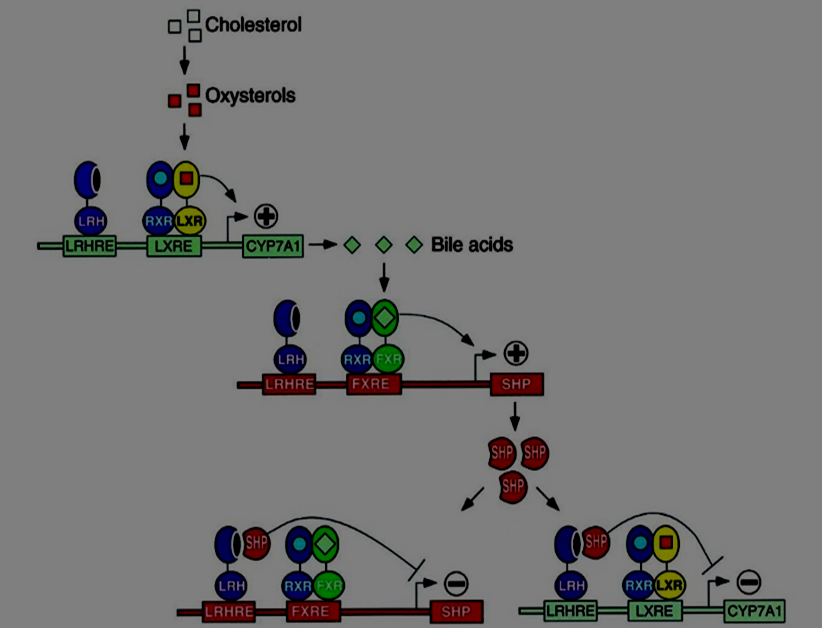
Figure 3 – A BAXS System regulatory subnetwork. Credit: [46]
2. Methods
2.1. About how the knowledge is gathered and organized
Figure 1 shows that each level of exploration provides new objects accompanied by the corresponding complementary information. Once the requested iteration levels are completed, proceeds the definition of possible PubMed IDs, which are related to the names and synonyms of the objects found (see PubMed IDs searching, Figure 1 and 2). Then, the abstracts related to those PubMed IDs are downloaded and a general summary is obtained, containing only sentences related to regulatory events (see abstracts downloading and summarizer steps, in Figure 1 and 2). Once the general summary is defined, a KB of regulatory events of the style described in Table 1 is available. As the figures show the KB is delivered to an inference engine, equipped with appropriate scripts, which makes possible the deduction of possible regulation pathways and regulatory subnets. Such pathways, ideally, should show paths ranging from the cell membrane (region near to the ligands), to the nuclear membrane (region near to the transcription factors). Figure 1 shows that the information collected, for the different objects of the network, is exported to prolog [51] format. This step shapes the different knowledge bases required for inferential analysis processes that we ran using prolog (so far). Other inference engines will be added as the queries and knowledge bases grow in time.
Table 2, numeral 1, declares that the CYP7A1 identifier corresponds to the human CYP7A1 protein and offers a list of synonyms accepted by the community for it. The first fact corresponds to the first thing that MESH says about CYP7A1. The second fact in numeral 1, corresponds to naming and synonyms information coming from HGNC and other consulting services. Numeral 2, in the same table, describes the identity tree that MESH currently offers for the CYP7A1 identifier; which makes possible to establish, for example, that CYP7A1 is a protein belonging to the family of hydrolases and the family of oxidases. Numeral 3, on the other hand, lists the molecular functions, biological processes, cellular components and possible relationships between them; and all of them correspond to the structure of the GeneOntology trees defined for CYP7A1. The lower part of numeral 3 shows relations of the type is_a, which expresses, for example, when one molecular function is a special case of another or when a molecular function is part of a biological process. These types of statements correspond to the branches of the trees to which we are accustomed in the MESH or GeneOntology web portals. The logical representation of such trees is achieved here by the logical representation of their branches, so their reconstruction only requires their logical reconnection. Finally, numeral 4, in Table 2, lists those logical facts required to explore regulatory pathways. In this case, it is required to establish which objects in the network are proteins, enzymes, transcription factors, receptors, ligands, among others. This is achieved by consulting the identity of each object in the network. We can ask, for instance: is CYP7A1 a member of the proteins family? The system can conclude that by reviewing the tree described in 2) and reflect, as a consequence, such a fact in 4).
Table 1 – A subset of regulatory events in the BAXS System GRN (manually defined)
|
base([ event('cholesterol',regulate,'oxysterols'), event('oxysterols',bind,'LXRa'), event('oxysterols',activate,'LXRa'), event('LXRa',associate,'RXR'), event('LXRa',bind,'LXRE'), event('LXRa',activate,'CYP7A1'), event('CYP7A1',increase,'ba'), event('ba',bind,'FXR'), event('ba',activate,'FXR'), event('FXR',associate,'RXR'), event('FXR',bind,'FXRE'), event('FXR',regulate,'SHP'), event('SHP',associate,'LRH'), event('LRH',bind,'LRHRE'), event('LRH',inhibit,'SHP'), event('LRH',inhibit,'CYP7A1') ]). |
The logical facts listed in 4), Table 2, depends on the satisfaction from objects, of specific restrictions related to specific queries, and they are deduced running internal inferential analysis. For instance, if we are searching a regulation pathway in which the possible transcription factors must be part of a specific biological process, then we must consult in the KBs which of them are available. Those conclusions must then be represented in 4). At this point we want to underline that all the information described above is automatically gathered and translated to logical format, and the purpose of this is to support (in time) a flexible framework in which to run flexible inferential analysis processes.
According to Figure 2, the first set of possible regulatory objects for a transcript under consideration includes: 1) An optional set of homologue obtained from Blast [49]; 2) the transcription factors (FTs), proposed using TFBIND [50]; and 3) the set of objects that could be provided by the person who models (the expert's objects). Blast allows to locate biological objects evolutionarily related to the sequence under study. TFBIND allows us to identify possible transcription factors, capable of anchoring to response elements (motifs) present in the DNA sequence provided. For each of such objects a process is run to define: a) its name and synonyms, b) Mesh ontology, and c) the related GeneOntology trees. This can be seen in the first three steps in Figure 2; the rest of the processes of this figure correspond to the rest of the exploratory levels indicated in Figure 1, and allows to explore new objects for the network using consulting services provided by PDB. Figure 1 illustrates the process of building the network as it proceeds from the cell nucleus to the cell membrane. Figure 2 describes the computer process that automatically builds the network of regulatory events, names and ontologies for the GRN's objects, following the biological path illustrated in Figure 1.
2.2. About the pathways and subnetworks searching
In this work, the search for regulatory pathways is guided by simple definitions like this: a regulation pathway is one in which a ligand recognizes a receptor by activating it, which triggers a cascade of regulatory events in which different types of proteins could be involved; event cascade that is closed by an event in which a transcription factor recognizes a response element, therefore activating, or inhibiting, the transcription of a target product (RNA or protein). In our framework, the solution finding consists in determining a collection of regulatory events that satisfy the restrictions implied by such a definition. Having knowledge such as the declared in Table 2, events that satisfy a set of specific restrictions can be explored in a KB like the one listed in Table 1. That is the type of heuristics that we follow here. Our goal is to develop a logical framework in which it may be possible to receive definitions like the one before and to construct knowledge bases richer enough to assist in their resolutions.
This work is also motivated about the searching of possible subnets of closely linked objects. Figure 3 shows an example from the BAXS system. There we see two main proteins, CYP7A1 and SHP. To discover subnets like the one shown, our approach is guided by the determination of regulation pathways which are connected through linking events; offering to who explores the network, possible linking alternatives between interesting pathways. The main idea is to determine subnets in which each of the proteins of interest are stimulated and inhibited. For example, if a subnet is found in which CYP7A1 is stimulated (first) and inhibited (later), and it is possible to observe in it some other protein that mediates between both scenarios, then other subnets could be determined, for such mediating protein, which show the conditions that rules its stimulation or inhibition. In Figure 3, CYP7A1 is stimulated and inhibited depending on the presence or absence of SHP, therefore it is important to find subnets in which SHP is stimulated or inhibited. It should be noted that two subnets coexist in Figure 3. One that shows the conditions that stimulate and inhibit the presence of CYP7A1, and another that describes the same for SHP. This work offers an automatic scanning option of possible subnets for each protein pointed above. Then, we must proceed (manually so far), to the analyzing of such subnets searching for a relationship of the style described in Figure 3. What follows includes a summary of the manual procedure to follow.
3. Results
The automatic modeling of the BAXS' system GRN proposed here is based on a list of proteins and various ligands linked to them. The proteins chosen correspond to a set of pathways modeled manually in [47] (see Table 3). The aim, in this case, is to take these proteins and develop an independent modeling process for each one. Here we focused on how modeling processes run for two of them: CYP7A1 and SHP. In the modeling process developed for each protein, the following data set were provided: 1) homologous proteins, 2) DNA sequence regulating region, 3) a list of possible regulating biological objects (ligands, proteins), and 4) an optional list of PubMed IDs, related with the GRN under modeling. These informational items were delivered to the pipeline described in Figures 1 and 2, producing a modeling plane for each protein; modeling planes that were automatically integrated. The general result of the modeling process consists of five knowledge bases, as described in Tables 1 and 2, and a collection of summarized abstracts related with the proteins on consideration.
Two general steps must be followed in order to find subnets as the one shown in Figure 3. The first step involves the search for pathways for the objects included in such a subnetwork, namely: bile acid, cholesterol, oxysterols, CYP7A1, LRH/NR5A2/FTF, LXR/NR1H2, RXRa, FXR/NR1H4 and SHP/NR0B2. The search for pathways for CYP7A1 and SHP were performed indicating to the system that they could start with one of the ligands or receptors provided, and be closed with some of the CYP7A1 and SHP/NR0B2 proteins.
Table 4 shows the kind of results obtained in the pathways searching (see pathways.txt, in supplemental material, for a full list of the inferred pathways). The result obtained in this first analysis step (Table 3), corresponds to the Regulation Patterns output, indicated in Figure 1 and Figure 2. Table 3 corresponds to a first set of results to compare a manual model with a semiautomatic modeling of pathways. The manual model described in [47] was based on PubMed references up until 2009; however, for the other model, we decided to do a general search in PubMed, in order to establish the pathways that the system is able to produce for the same proteins listed in the original work. It can be seen that the system does not retrieve all the references described manually, but offers alternative descriptions for the pathways under consideration. It is clear now that some improvements are required. The simple subject-verb-object parsing that we use cannot capture all possibilities about regulatory sentences. An example of this is: "Moreover, direct regulation of NRF2 by AHR contributes to couple phase I and II enzymes into an integrated system facilitating more effective xenobiotic and carcinogen detoxification" (PMID: 15790560). In this case, the event 'AHR',regulate,'NRF2' should be detected but it is not. However, as we will see in the following sections, our system produces data for alternative analysis.
About the NRF2 protein, the system automatically proposes 12 pathways and produces the documentation for the different events that shape them. Once the accuracy of the modeling of each event has been manually validated as type P (Positive), F (False) or U (added by the user), the knowledge base can be automatically updated. In this case, 42 sentences are listed as supporting the 12 pathways found. For the 42 sentences linked to the different events, 25 turn out to be correctly modeled, 17 are not, and 5 new ones are identified by the user. This procedure reduces the 12 original pathways to 4. Table 3 shows only one of them, pointing out the NRF2's regulation mediated by AHR. We highlight here that the manual analysis of the 3 remaining pathways allows us detecting that there is mutual regulation between NRF2 and AHR. The following sentence remarks it: "Conversely, this study demonstrates that NRF2 regulates expression of Ahr and subsequently modulates several downstream events of the Ahr signaling cascade, including..." (PMID: 17709388). Details for both, the tagging of the original 42 sentences and the final 4 pathways for NRF2, can be seen in eventsDocNRF2.txt and pathwaysDocNRF2.txt (see supplemental material). An analysis like the one above for the whole list of proteins in Table 3, defines the way we are proceeding to evaluate the accuracy of the system about the modeling of regulatory events. In the case of pathwaysDocNRF2.txt the automatically generated documentation style is the one depicted in Table 4. Such a table illustrates how the system helps finding subnets inside a network describing proteins with feedback regulation behaviors. This will be explained in the following paragraphs.
Our second experimental analysis step involves the searching of subnetworks. In this case, we follow an analytic procedure supported by: a) the knowledge bases of the style described in Tables 1 and 2 (see details in kBase.pl, minedObjects.pl, ontologyMESH.pl, ontologyGO.pl and pathwaysObjects.pl, from the supplemental material); b) the compilation of regulatory sentences obtained from the abstracts downloaded (see kBaseDoc.txt, supplemental material); and c), the collection of subnetworks automatically defined for CYP7A1 and SHP/NR0B2 (see CYP7A1_chainsPathways.txt, NR0B2_chainsPathways.txt, supplemental material). For example, Table 1 contains the knowledge base corresponding to the subnet shown in Figure 3 (manually modeled) (see kBase.pl for the automatic version), and it shows that the event event ('CYP7A1', increase, 'ba') links an upper pathway with an intermediate one. If the system supplies such linking events, as part of subnetwork alternatives composed of three pathways (upper, intermediate and lower) (see Table 5, for instance); then it could be possible to explore and to choose subnets from them. The semi automatic procedure followed here to explore and define a possible regulation subnet for a pair of proteins can be summarized as follows:
Table 2 – A sample of knowledge representation for objects in the BAXS' System GRN
|
1) Names, synonyms and retrieved basic facts
cyp7a1_protein_human('CYP7A1').
synonyms('CYP7A1', [ 'cytochrome P450 family 7 subfamily A member 1', 'CYP7', 'Cytochrome P450 7A1', .... ]).
nr1h3_protein_mouse('NR1H3').
synonyms('NR1H3',[ 'LXR-a', 'RLD-1','LXRa', 'Oxysterols receptor LXR-alpha', 'Liver X receptor alpha', 'Nuclear receptor subfamily 1 group H member 3', .... ]).
.. |
2) MESH ontology
object('CYP7A1'). is_a('CYP7A1','CYP7A1 protein, human'). is_a('CYP7A1 protein, human','Cholesterol 7-alpha-Hydroxylase'). is_a('Cholesterol 7-alpha-Hydroxylase','Cytochrome P450 Family 7'). is_a('Cytochrome P450 Family 7','Cytochrome P-450 Enzyme System'). is_a('Cytochrome P-450 Enzyme System','Cytochromes'). is_a('Cytochromes','Enzymes and Coenzymes'). is_a('Cytochromes','Hemeproteins'). is_a('Hemeproteins','Proteins'). is_a('Cytochrome P-450 Enzyme System','Mixed Function Oxygenases'). is_a('Mixed Function Oxygenases','Oxygenases'). is_a('Oxygenases','Oxidoreductases'). is_a('Oxidoreductases','Enzymes'). is_a('Cholesterol 7-alpha-Hydroxylase','Steroid Hydroxylases'). is_a('Steroid Hydroxylases','Cytochrome P-450 Enzyme System').
object('NR1H3'). .. .. |
|
3) GeneOntology trees
mf('CYP7A1',[ 'cholesterol 7-alpha-monooxygenase activity', 'heme binding', 'iron ion binding' ]).
bp('CYP7A1',[ 'bile acid biosynthetic process', 'cellular response to cholesterol', 'sterol metabolic process', .... ]).
cc('CYP7A1',[ 'endoplasmic reticulum membrane', 'intracellular membrane-bounded organelle', 'organelle membrane' ]).
is_a( 'cholesterol 7-alpha-monooxygenase activity', 'steroid hydroxylase activity' ).
is_a( 'steroid hydroxylase activity','monooxygenase activity' ).
.. |
4) Logical facts inferred from 1, 2 and 3 (improved manually)
protein('CYP7A1'). enzyme('CYP7A1'). receptor('NR1H4'). protein('NR1H4'). receptor('SHP'). protein('SHP'). ligand('bile acid'). ligand('oxysterols'). ligand('cholesterol'). receptor('RXR'). protein('RXR'). receptor('NR5A2'). protein('NR5A2'). receptor('LRH'). protein('LRH'). receptor('NR1H3'). protein('NR1H3'). receptor('NR1H2'). protein('NR1H2'). transcription_factor('RUNX1'). protein('RUNX1'). transcription_factor('PBX1'). protein('PBX1'). transcription_factor('CLIP1'). protein('CLIP1'). protein('CLIP2'). ligand('CLIP3'). protein('CLIP3'). enzyme('BAAT'). transcription_factor('CAP'). .. |
1) Using the files mentioned above we follow this procedure: a) based on pathways.txt we choose a particularly interesting pathway; one that relates CYP7A1 and oxysterols and involves an interesting transcriptional factor working on it (see the one in Table 4(a), upper side). Then, we use the pathway chosen to run an automatic search for possible subnetworks relating the CYP7A1 and SHP/NR0B2 proteins; this step defines the contents of CYP7A1_chainsPathways.txt. b) We explore the CYP7A1_subnetworks.txt file to find an interesting subnetwork; let's say that we select one that shows a linking event indicating that CYP7A1 regulates bile acid (see Table 4(a) lower side and Table 5(a) upper side). c) We continue the manual CYP7A1's subnetworks exploration, looking for an intermediate pathway that could involve the SHP/NR0B2 transcriptional stimulation, mediated by the bile acid ligand; this lead us to the pathway depicted in Table 4(b) upper side and Table 5(a) middle side. And finally d), we continue the exploration of CYP7A1's subnetworks searching for a second linking event that could guide us to a last pathway that could inhibit CYP7A1's transcription mediated by SHP/NR0B2; this lead us to the linking event described in Table 4(b) and Table 5(a) lower side (see subnetwork 9/54). The second linking event is defined by means of some criteria chosen by the user, in this case the transcription factor involved (LRH/NR5A2). At this point we have a subnetwork that could regulate CYP7A1's transcription mediated by SHP/NR0B2 (Table 5 (a)).
2) Once we define the subnet above, we wonder if there will be a subnet that regulates the inhibition of NR0B2, mediated by the same linking event that regulates CYP7A1's inhibition. We are also interested in determining if NR0B2 is capable of inhibiting itself. To explore this possibility we use the middle pathway in Table 5 (a) and use the system to automatically define subnetworks for SHP/NR0B2 based on it; this step defines the content of the NR0B2_chainsPathways.txt file. Since we want the same last CYP7A1's linking event event ('NR0B2', bind, 'NR5A2'), and NR0B2 inhibiting itself, then the closing pathway for NR0B2's inhibition could look like: 'NR0B2',bind,'NR5A2';'NR5A2',inhibit,'NR0B2';'NR0B2',inhibit,'NR0B2'. And because NR0B2 must mediate its inhibition by itself, the subnetwork must have only 2 pathways. With these restrictions guiding the searching we define the events described in Table 5 (b) (see subnetwork 19/92). The sentences in Table 4 were defined pairing the events in the pathways with the contents of kBaseDoc.txt, having in mind different synonyms for regulate, inhibit, associate and bind; for instance, regulate could mean induce, increase, control, activate and so on (see relations-functions.txt, supplementary material).
Table 3 – Comparative modeling of pathways to guide the modeling and analysis processes. Credit: [47] *
|
Regulation pathways |
PUBMED IDs (Manual)* |
PUBMED IDs (Semi-Automatic) |
|
CYP7A1 synthesis through (oxysterol-LXR)-(RXR)-LXRE |
11030331 |
18782758, 22235657, 11030331** |
|
CYP7A1 inhibition through (SHP-LRH) |
11030331 |
15976031, 18385139 , 11030331** |
|
SHP synthesis stimulation through (FXR-ba)-RXR |
11030331 |
22094889, 14699511, 11030331** |
|
SHP synthesis inhibition through (SHP-LRH) |
11030331 |
15976031, 11518759 , 11030331** |
|
Cyp2b6 synthesis through (PB-CAR)-(RXR)-DR4 |
12573483, 15000748 |
16725103, 12573483**, 15000748** |
|
BSEP synthesis through (FXR-ba)-RXRalpha-IR1 |
11509573, 14699511 |
11509573**, 14699511** |
|
ASBT synthesis through (PPARalpha)-(HNF1alpha) |
12055195 |
23991366, 30105244 |
|
CYP2B10 synthesis through PB-(CAR-RXR)-DR1 |
12573483 |
16725103, 12573483** |
|
CYP2D6 synthesis through HNF4(alpha)-DR1 |
8810289 |
22383578, 8810289**, 11723233 |
|
HNF1(alpha) synthesis through RXR(alpha)-RA-DR1 |
11027556 |
11589627, 11027556** |
|
HNF4(alpha) synthesis through RXR(alpha)-RA-DR1 |
11027556 |
11589627, 11027556** |
|
I-BABP synthesis through (PPAR(alpha)-cholesterol)-RXR) |
15936983 |
14769039, 15103326, 15936983** |
|
MRP2 synthesis through (CAR-pb)-RXR(alpha) |
12573483, 19638083 |
11113125, 15832810, 11706036 27155371, 15832810, 11706036 |
|
MRP2 synthesis through (PPAR)-RXR(alpha) |
15936983, 30105244 |
9744281, 19588995, 30105244** |
|
MRP2 synthesis through (RXR(alpha-RAR(alpha)) |
12145812 |
10979971, 12145812** |
|
OATP8 synthesis through (FXR-cda)-RXR |
12055601 |
11706036, 12055601** |
|
MDR3 synthesis through (FXR-cda)-RXR(alpha) |
14527955 |
11706036, 16706681 |
|
PLC(gamma)1 synthesis through (VDR-RAR)-DR6 |
12520529 |
12899516, 12520529** |
|
MRP3 synthesis through (VDR-9cRA)-RXR(alpha) |
15824121 |
15824121** |
|
MRP3 synthesis through (VDR-LCA)-RXR(alpha) |
15824121 |
15824121** |
|
MRP3 synthesis through (VDR-VDR3)-RXR(alpha) |
15824121 |
15824121** |
|
Nfr2 synthesis through (Ahr-TCDD) |
15790560 |
19456125, 24914470 |
Note: *It coincides with the other method (18/21).
Table 4 – The list of pathways produced by the subnetworks finding procedure
|
a) Pathway 1:
'oxysterols',bind,'NR1H2'; 'NR1H2',regulate,'CYP7A1'; 'oxysterols',regulate,'CYP7A1'
'oxysterols',bind,'NR1H2': When cholesterol accumulates, oxysterols bind to LXRbeta, and the LXRbeta/RXR complex dissociates from ABCA1, restoring ABCA1 activity and allowing apoA-I-dependent cholesterol secretion. PMID: 18782758. 'NR1H2',regulate,'CYP7A1': The main activator is LXR (in rodents), increasing CYP7A1 transcriptional activity. PMID: 22235657. 'oxysterols',regulate,'CYP7A1': The catabolism of cholesterol into bile acids is regulated by oxysterols and bile acids, which induce or repress transcription of the pathway's rate-limiting enzyme cholesterol 7alpha-hydroxylase (CYP7A1). PMID: 11030331. Linking event: 'CYP7A1',regulate,'bile acid': The rate of bile acid synthesis is controlled by the activity of the enzyme, cholesterol 7 alpha-hydroxylase, encoded on the CYP7A1 gene. PMID: 15769658. |
b) Pathway 2:
'bile acid',bind,'NR1H4'; 'NR1H4',regulate,'NR0B2'; 'bile acid',regulate,'NR0B2'
'bile acid',bind,'NR1H4': Conformational changes induced by bile acid binding to pre-bound FXR leads to increased expression of a variety of genes. PMID: 22094889.
'NR1H4',regulate,'NR0B2': A key regulator of hepatocellular bile salt homeostasis is the bile acid receptor/farnesoid X receptor FXR, which activates transcription of the BSEP and OATP8 genes and of the small heterodimer partner 1 (SHP). PMID: 14699511.
'bile acid',regulate,'NR0B2': Feedback repression of CYP7A1 is accomplished by the binding of bile acids to FXR, which leads to transcription of SHP. PMID: 11030331.
Linking event: 'NR0B2',bind,'NR5A2': The functional interaction between the orphan nuclear receptors small heterodimer partner (SHP) and liver receptor homolog 1 (LRH-1), where SHP binds to LRH-1 and represses its constitutive transcriptional activity, is crucial for regulating genes involved in cholesterol homeostasis. PMID: 15976031. |
|
c) Pathway 3:
'NR0B2',bind,'NR5A2'; 'NR5A2',inhibit,'CYP7A1'; 'NR0B2',inhibit,'CYP7A1'
'NR5A2',inhibit,'CYP7A1': These data suggest that PGC-1alpha is an important co-activator for LRH-1 and that SHP targets the interaction between LRH-1 and PGC-1alpha to inhibit CYP7A1 expression. PMID: 18385139.
'NR0B2',inhibit,'CYP7A1': Elevated SHP protein then inactivates LRH-1 by forming a heterodimeric complex that leads to promoter-specific repression of both CYP7A1 and SHP. PMID: 11030331. |
d) Pathway 4:
'NR0B2',bind,'NR5A2'; 'NR5A2',inhibit,'NR0B2'; 'NR0B2',inhibit,'NR0B2'
'NR5A2',inhibit,'NR0B2': Results revealed that FTF was a dominant negative factor that was induced by bile acid-activated FXR to inhibit both CYP7A1 and SHP transcription. PMID: 11518759.
'NR0B2',inhibit,'NR0B2': Elevated SHP protein then inactivates LRH-1 by forming a heterodimeric complex that leads to promoter-specific repression of both CYP7A1 and SHP. PMID: 11030331. |
At the moment we can enumerate the following results: the automatic generation of knowledge bases on regulatory events; the gathering of detailed information for each of the objects provided by the user; the automatic addition of possible new objects to a model of a network; access to web services based on ontologies for consultation and consequent local organization of knowledge bases for an object (identity, from Mesh, and molecular function, biological processes and cellular component, from GeneOntology); automatic consultation and download of abstracts, duly related to a GRN on modeling; the generation of diverse knowledge bases in prolog format, immediately available for consulting and inference; the integration of a NLP tool for the processing of abstracts; and the programming and testing of various archetypal queries for the analysis of a network. Table 3 shows that it is possible to discover pathways that, duly analyzed by a user, are consistent and support those that were defined manually. In this regard, most of the pathways listed in Table 3, include references to PubMed IDs different from those manually provided; therefore, offering references to alternative works that support the same pathways. Tables 4 and 5 show that the system allows to define subnets and also provides the corresponding linking events. At this end, the system automatically integrates the results of independent experiments for CYP7A1 and NR0B2, which generates ontologies and knowledge bases, which allow rediscovering the network shown in Figure 3. The automatic modeling and integration of the results obtained for the proteins CYP7A1 and NR0B2, has allowed us to propose a semi automatic analysis procedure to assist in the discovery of subnetworks inside a general GRN.
Table 5 – The subnetworks manually defined using the knowledge automatically gathered *
|
a) Subnetwork for CYP7A1's regulation ------------------------------------------------- Pathway 1 => 'oxysterols',bind,'NR1H2'; 'NR1H2',regulate,'CYP7A1'; 'oxysterols',regulate,'CYP7A1'
Possible linking events: 'CYP7A1',regulate,'bile acid'; (..… other possible linking events….);
Pathway 2 => 'bile acid',bind,'NR1H4'; 'NR1H4',regulate,'NR0B2'; 'bile acid',regulate,'NR0B2'
Possible linking events: 'NR0B2',bind,'NR5A2'
Pathway 3=> 'NR0B2',bind,'NR5A2'; 'NR5A2',inhibit,'CYP7A1'; 'NR0B2',inhibit,'CYP7A1' ------------------------------------------------ |
b) Subnetwork for NR0B2's regulation
------------------------------------------------- Pathway=> 2 'bile acid',bind,'NR1H4'; 'NR1H4',regulate,'NR0B2'; 'bile acid',regulate,'NR0B2'
Linking events: 'NR0B2',bind,'NR5A2'
Pathway=> 4 'NR0B2',bind,'NR5A2'; 'NR5A2',inhibit,'NR0B2'; 'NR0B2',inhibit,'NR0B2' ------------------------------------------------- |
Note: *For a quick parsing of subnetworks, use the pathway 2 in this format:
'bile acid',bind,'NR1H4';'NR1H4',regulate,'NR0B2';'bile acid',regulate,'NR0B2'
Conclusions and Further Work
The system described here integrates different sources of information, from human provided specifications and hints to ontological knowledge represented in databases in the Internet; including information written in human natural languages in papers that are automatically processed and transformed into supporting knowledge bases. For example, the conceptual trees built for each object in a GRN, consulted from Mesh and GeneOntology, define local formatted conceptual trees that assist the automatic and semiautomatic exploration of pathways and subnets. Only conceptual trees (based on Mesh services) are being exploited at the moment; the remaining trees (molecular function, biological processes, and cellular component) must be incorporated in future work, using new user defined constraints that guide the system's exploration facilities supported in them. The automatic definition of computational representations of the mentioned trees as local ontologies, is achieved for each object for a network under construction, and they are already successfully integrated in exploration tasks. It is necessary to improve both the way in which regulatory events are structured and the quality of the regulation pathways that are obtained by the inference processes. This task has to do with the natural language processing of the abstracts and that must be improved too. Search spaces, both pathway and subnet, can be large since the KBs involved can contain many elements. Therefore, meta-heuristics that help the user to explore those spaces in a more friendly way should be investigated. At the moment the user can tag the events that shape a set of inferred pathways and manually improve the quality of the knowledge base about them; this consequently improves the quality of the pathways and subnetworks related to them. About the subnetworks searching we have explored and illustrated how to discover a subnetwork inside a bigger GRN; such a semiautomatic procedure must be assisted by graphical options which are not available yet. Some heuristics are also required to restrict the new objects from the PDB proposed for a network, in order to keep them close to the profile of those that have already been proposed by the user/modeler, or have been already included in a previous modeling layer of the network being built. The use of statistical measures is necessary to assess the quality of the results presented so far. Table 3 is offered as proof of the concept as the system was able to recover all the pathways that were identified by humans in a previous manual modeling experiment. However, those experiments differ in time frames and the system does not recover all the previous pathways from the same papers (in 3 out of 21 attempts). We plan to device a more precise statistical analysis over alternative outputs produced by the system in future work.
Supplementary materials
The supplementary material that supports this paper can be downloaded from: https://doi.org/10.13140/RG.2.2.26897.86884.
Acknowledgement
We appreciate the support provided to us by our universities for the development of this project.
|
Conflict of Interest |
Конфликт интересов |
|
None declared. |
Не указан. |
Список литературы
Rodchenkov I. Pathway Commons 2019 Update: integration, analysis and exploration of pathway data / Rodchenkov I. et al. //Nucleic Acids Res. -2020. -Vol. 48. -No. issue_D1. -P. D489-D497. -URL:https://doi.org/10.1093/nar/gkz946
Croft D. Reactome: a database of reactions, pathways and biological processes / Croft D. et al. //Nucleic Acids Res. -2011. -Vol. 39. -No issue_1. -P. D691-D697. -URL:https://doi.org/10.1093/nar/gkq1018
Sirin E. Pellet: A practical OWL-DL reasoner / Sirin E. et al. //Journal of Web Semantics. -2007. -Vol. -No. issue_2. -P. 51-53. -URL:https://doi.org/10.1016/j.websem.2007.03.004
Munoz-Torres M. Get GO! Retrieving GO Data Using AmiGO, QuickGO, API, Files, and Tools / Munoz-Torres M., Carbon S. //Methods Mol Biol. -2017. -Vol. 1446. -P.149-160. -URL:https://doi.org/10.1007/978-1-4939-3743-1_11
Khamparia A. Comprehensive analysis of semantic web reasoners and tools: a survey / Khamparia, A., Pandey, B. //Educ Inf Technol. -2017. -Vol. 22. -P. 3121–3145. -URL:https://doi.org/10.1007/s10639-017-9574-5
Dickerson J.A. Creating Metabolic Network Models using Text Mining and Expert Knowledge / Dickerson J.A. et al. //Computational Biology and Genome Informatics.: Edited by World Scientific Publishing. -2003. -P. 268. -URL:https://doi.org/10.1142/5183
Chen H. Content-rich biological network constructed by mining PubMed abstracts / Chen H., Sharp B.M. //BMC Bioinformatics. -2004. -Vol. 5. -No. 147. -URL:https://doi.org/10.1186/1471-2105-5-147
Li S. Constructing biological networks through combined literature mining and microarray analysis: a LMMA approach / Li S., Wu L., Zhang Z. //Bioinformatics. -2006. -Vol. 22. -No issue_17. -P. 2143-2150. -URL:https://doi.org/10.1093/bioinformatics/btl363
Gevaert O. A framework for elucidating regulatory networks based on prior information and expression data / Gevaert O., Van Vooren S., De Moor B. //Ann N Y Acad Sci. -2007. -Vol. 1115. -No issue_1. -P. 240-248. -URL:https://doi.org/10.1196/annals.1407.002
Steele E. Literature-based priors for gene regulatory networks / Steele E. et al. //Bioinformatics. -2009. -Vol. 25. -No. 14. P. 1768-1774. -URL:https://doi.org/10.1093/bioinformatics/btp277
Zhu F. Biomedical text mining and its applications in cancer research / Zhu F. et al. //J Biomed Inform. -2013. -Vol. 46. -No. issue_2. -P. 200-211. -URL:https://doi.org/10.1016/j.jbi.2012.10.007
Žitnik S. Sieve-based relation extraction of gene regulatory networks from biological literature / Žitnik S. //BMC Bioinformatics. -2015. -Vol. 16. -No. suppl_16. -P. S1. DOI: -URL:https://doi.org/10.1186/1471-2105-16-S16-S1
Bouaziz J. How Artificial Intelligence Can Improve Our Understanding of the Genes Associated with Endometriosis: Natural Language Processing of the PubMed Database / Bouaziz J. et al //Biomed Res Int. -2018. -No. 6217812. -URL:https://doi.org/10.1155/2018/6217812
Baral C. A knowledge based approach for representing and reasoning about signaling networks / Baral C. et al. //Bioinformatics. -2004. -Vol. 20. -No. suppl_1. -P. i15-i22. -URL:https://doi.org/10.1093/bioinformatics/bth918
Tran N. Knowledge-based framework for hypothesis formation in biochemical networks / Tran N. et al. //Bioinformatics. -2005. -Vol. 21 -No. suppl_2. P. ii213-ii219. -URL:https://doi.org/10.1093/bioinformatics/bti1134
Muggleton S.H. Machine Learning for Systems Biology / Muggleton S.H. //Inductive Logic Programming, Lecture Notes in Computer Science: Edited by Kramer S., Pfahringer B., Springer. -2005. -Vol. 3625. -P. 416-423. -URL:https://doi.org/10.1007/11536314_27
Tamaddoni-Nezhad A. Application of abductive ILP to learning metabolic network inhibition from temporal data / Tamaddoni-Nezhad A. et al. //Machine Learning. -2006. Vol. 64. -P. 209–230. -URL:https://doi.org/10.1007/s10994-006-8988-x
Muggleton S. Knowledge Mining Biological Network Models / Muggleton S. //6th IFIP TC 12 International Conference on Intelligent Information Processing (IIP). -2010. -P. 2-2. -URL:https://doi.org/10.1007/978-3-642-16327-2_2
Linde J. Data- and knowledge-based modeling of gene regulatory networks: an update / Linde J. et al. //EXCLI J. -2015. -Vol. 14. -P. 346-378. -URL:https://doi.org/10.17179/excli2015-168
Rougny A. A logic-based method to build signaling networks and propose experimental plans / Rougny A. et al.. //Sci Rep. -2018. -Vol. 8. -No. 7830. -URL:https://doi.org/10.1038/s41598-018-26006-2
Kitano H. Using process diagrams for the graphical representation of biological networks / Kitano H. et al. //Nat Biotechnol. -2005. -Vol. 23. -No. 8. -P. 961-966. -URL:https://doi.org/10.1038/nbt1111
Kitano H. Accelerating systems biology research and its real world deployment. //NPJ Syst Biol Appl. -2015. -Vol. 1. -No. 15009. -URL:https://doi.org/10.1038/npjsba.2015.9
Kitano H. Artificial Intelligence to Win the Nobel Prize and Beyond: Creating the Engine for Scientific Discovery / Kitano H. //AI Mag. -2016. -Vol. 37. -No. 1. -P. 39-49. -URL:https://doi.org/10.1609/aimag.v37i1.2642
Hsin K.Y. systemsDock: a web server for network pharmacology-based prediction and analysis / Hsin K.Y. et al. //Nucleic Acids Res. -2016. -Vol. 44. -No issue_W1. -P W507-W513. -URL:https://doi.org/10.1093/nar/gkw335
Emmert-Streib F., Dehmer M.,, Haibe-Kains B. Gene regulatory networks and their applications: understanding biological and medical problems in terms of networks / Emmert-Streib F., Dehmer M.,, Haibe-Kains B. //Front Cell Dev Biol. -2014. -Vol. 2. -No. 38. -URL:https://doi.org/10.3389/fcell.2014.00038
Demir E. The BioPAX community standard for pathway data sharing / Demir E. et al. //Nat Biotechnol. -2010. -Vol. 28. -No 9. P. 935–942. -URL: https://doi.org/10.1038/nbt.1666
Musen M.A. The Protégé Project: A Look Back and a Look Forward / Musen M.A et al. //AI Matters. -2015. -Vol. 1. -No. 4.. -URL:https://doi.org/10.1145/2757001.2757003
Fiorini N. Towards PubMed 2.0 / Fiorini N., Lipman D. J., Lu Z. //eLife. -2017. -Vol. 6. -No e28801. -URL:https://doi.org/10.7554/eLife.28801
Rose P.W. The RCSB protein data bank: integrative view of protein, gene and 3D structural information / Rose P.W. et al. //Nucleic Acids Res. -2017. -Vol. 45. -No. issue_D1. -P. D271-D281. -URL:https://doi.org/10.1093/nar/gkw1000
Berman H. M. The Protein Data Bank archive as an open data resource / Berman H. M. et al //J Comput Aided Mol Des. -2014. -Vol. 28. -No. 10. -P. 1009-1014. URL:https://doi.org/10.1007/s10822-014-9770-y
Gray K.A. A review of the new HGNC gene family resource / Gray K.A. et al. //Hum Genomics. -2016. -Vol. 10. -No. 6. -URL:https://doi.org/10.1186/s40246-016-0062-6
Thomas P.D. The Gene Ontology and the Meaning of Biological Function / Thomas P.D. //Methods Mol Biol. -2017. -Vol. 1446. -P. 15-24. -URL:https://doi.org/10.1007/978-1-4939-3743-1_2
Pundir S. UniProt Protein Knowledgebase / Pundir S, Martin MJ, O'Donovan C. //Methods Mol Biol. -2017. Vol. 1558. P. 41-55. -URL:https://doi.org/10.1007/978-1-4939-6783-4_2
Baumann N. How to use the medical subject headings (MeSH) / Baumann N. //Int J Clin Pract. -2016. -Vol. 70. -No. issue_2. -P. 171-174. -URL:https://doi.org/10.1111/ijcp.12767
Bhalla U. S. Understanding complex signaling networks through models and metaphors / Bhalla U. S. //Prog Biophys Mol Biol. -2003. -Vol. 81. -No. issue_1. -P. 45-65. -URL:https://doi.org/10.1016/s0079-6107(02)00046-9
Shao Li, Lijiang Wu, Zhongqi Zhang (2006). Constructing biological networks through combined literature mining and microarray analysis: a LMMA approach/ Shao Li, Lijiang Wu, Zhongqi Zhang. //Bioinformatics. -2006. -Vol. 22. No issue_17. -P. 2143–2150, https://doi.org/10.1093/bioinformatics/btl363
Li X. Global mapping of gene/protein interactions in PubMed abstracts: a framework and an experiment with P53 interactions / Li X. et al. //J Biomed Inform. -2007. -Vol. 40. -No issue_5. -P. 453-464. -URL:https://doi.org/10.1016/j.jbi.2007.01.001
Cho K.H. Reverse engineering of gene regulatory networks / Cho K.H. et al. //IET Syst Biol. -2007. -Vol. 1. -No 3. -P. 149-163. -URL:https://doi.org/10.1049/iet-syb:20060075
Camacho D. Comparison of reverse-engineering methods using an in silico network / Camacho D. et al. //Ann N Y Acad Sci. -2007. -Vol. 1115. -No. issue_1. -P. 73-89. URL:https://doi.org/10.1196/annals.1407.006
Karlebach G. Modelling and analysis of gene regulatory networks / Karlebach G., Shamir R. //Nat Rev Mol Cell Biol. -2008. -Vol. 9. -No 10. -P. 770-780. -URL:https://doi.org/10.1038/nrm2503
Viswanathan G.A. Getting started in biological pathway construction and analysis / Viswanathan G.A. et al. //PLoS computational biology. -2008. -Vol. 4. -No. 2. -P. e16. -URL:https://doi.org/10.1371/journal.pcbi.0040016
Janky R. iRegulon: from a gene list to a gene regulatory network using large motif and track collections / Janky R. et al. //PLoS Comput Biol. -2014. -Vol. 10. No. 7. -URL:https://doi.org/10.1371/journal.pcbi.1003731
Cussat-Blanc S. Artificial Gene Regulatory Networks-A Review / Cussat-Blanc S., Harrington K., Banzhaf W. //Artif Life. -2018. -Vol. 24. -No issue_4. -P. 296-328. -URL:https://doi.org/10.1162/artl_a_00267
Dávila J. Una gramática de estilos para resumir textos en español [A grammar of style to summarize Spanish texts] / Dávila J., Contreras H. //Revista de La Sociedad Español para el Procesamiento del Lenguaje Natural (SEPLN). -2002. -Vol. 29. -P. 265-273. -URL:http://www.saber.ula.ve/handle/123456789/15963 [in Spanish]
Parra M.M. Un Modelo Computacional para la Generación de Resúmenes Automáticos de Artículos Científicos en Español [A computational model for the generation of automatic, Spanish, scientific abstracts] / Parra M.M., Dávila J. //ACTAS del IX Simposio Internacional de Comunicación Social. -2005. -P. 213-216. -URL:https://www.researchgate.net/publication/44476598 [in Spanish]
Lu TT. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors / Lu TT. et al. //Mol Cell. -2000. -Vol. 6. -No 3. -P. 507-515. -URL:https://doi.org/10.1016/s1097-2765(00)00050-2
Schmidt O. (2009). A standard-compliant global map of the bile acid/xenobiotic signalling network: Construction and automated query processing / Schmidt O. et al. //Proceedings of The 2009 International Conference on Bioinformatics and Computational Biology (BIOCOMP'2009). -2009. -Vol. II. -P. 657-663. -URL:https://www.researchgate.net/publication/344755794
Lee J. Processing SPARQL queries with regular expressions in RDF databases / Lee J. et al. //BMC Bioinformatics. -2011. -Vol. 12. -No suppl_2. -P. S6. -URL:https://doi.org/10.1186/1471-2105-12-S2-S6
Ladunga I. Finding homologs to nucleotide sequences using network BLAST searches / Ladunga I. //Curr Protoc Bioinformatics. -2002. ; -Vol 00. -No. issue_1. -P. 3.3.1-3.3.25. -URL:https://doi.org/10.1002/0471250953.bi0303s00
Tsunoda T., Takagi T. Estimating transcription factor bindability on DNA / Tsunoda T., Takagi T. //Bioinformatics. -1999. -Vol. 15. -No. issue_7. -P. 622-630. -URL:https://doi.org/10.1093/bioinformatics/15.7.622
Wielemaker J. SWI-prolog / Wielemaker, J. et al. //Theory and Practice of Logic Programming. -2012. -Vol. 12. -No 1-2. -P. 67–96. -URL:https://doi.org/10.1017/S1471068411000494
Список литературы
Rodchenkov I. Pathway Commons 2019 Update: integration, analysis and exploration of pathway data / Rodchenkov I. et al. //Nucleic Acids Res. -2020. -Vol. 48. -No. issue_D1. -P. D489-D497. -URL:https://doi.org/10.1093/nar/gkz946
Croft D. Reactome: a database of reactions, pathways and biological processes / Croft D. et al. //Nucleic Acids Res. -2011. -Vol. 39. -No issue_1. -P. D691-D697. -URL:https://doi.org/10.1093/nar/gkq1018
Sirin E. Pellet: A practical OWL-DL reasoner / Sirin E. et al. //Journal of Web Semantics. -2007. -Vol. -No. issue_2. -P. 51-53. -URL:https://doi.org/10.1016/j.websem.2007.03.004
Munoz-Torres M. Get GO! Retrieving GO Data Using AmiGO, QuickGO, API, Files, and Tools / Munoz-Torres M., Carbon S. //Methods Mol Biol. -2017. -Vol. 1446. -P.149-160. -URL:https://doi.org/10.1007/978-1-4939-3743-1_11
Khamparia A. Comprehensive analysis of semantic web reasoners and tools: a survey / Khamparia, A., Pandey, B. //Educ Inf Technol. -2017. -Vol. 22. -P. 3121–3145. -URL:https://doi.org/10.1007/s10639-017-9574-5
Dickerson J.A. Creating Metabolic Network Models using Text Mining and Expert Knowledge / Dickerson J.A. et al. //Computational Biology and Genome Informatics.: Edited by World Scientific Publishing. -2003. -P. 268. -URL:https://doi.org/10.1142/5183
Chen H. Content-rich biological network constructed by mining PubMed abstracts / Chen H., Sharp B.M. //BMC Bioinformatics. -2004. -Vol. 5. -No. 147. -URL:https://doi.org/10.1186/1471-2105-5-147
Li S. Constructing biological networks through combined literature mining and microarray analysis: a LMMA approach / Li S., Wu L., Zhang Z. //Bioinformatics. -2006. -Vol. 22. -No issue_17. -P. 2143-2150. -URL:https://doi.org/10.1093/bioinformatics/btl363
Gevaert O. A framework for elucidating regulatory networks based on prior information and expression data / Gevaert O., Van Vooren S., De Moor B. //Ann N Y Acad Sci. -2007. -Vol. 1115. -No issue_1. -P. 240-248. -URL:https://doi.org/10.1196/annals.1407.002
Steele E. Literature-based priors for gene regulatory networks / Steele E. et al. //Bioinformatics. -2009. -Vol. 25. -No. 14. P. 1768-1774. -URL:https://doi.org/10.1093/bioinformatics/btp277
Zhu F. Biomedical text mining and its applications in cancer research / Zhu F. et al. //J Biomed Inform. -2013. -Vol. 46. -No. issue_2. -P. 200-211. -URL:https://doi.org/10.1016/j.jbi.2012.10.007
Žitnik S. Sieve-based relation extraction of gene regulatory networks from biological literature / Žitnik S. //BMC Bioinformatics. -2015. -Vol. 16. -No. suppl_16. -P. S1. DOI: -URL:https://doi.org/10.1186/1471-2105-16-S16-S1
Bouaziz J. How Artificial Intelligence Can Improve Our Understanding of the Genes Associated with Endometriosis: Natural Language Processing of the PubMed Database / Bouaziz J. et al //Biomed Res Int. -2018. -No. 6217812. -URL:https://doi.org/10.1155/2018/6217812
Baral C. A knowledge based approach for representing and reasoning about signaling networks / Baral C. et al. //Bioinformatics. -2004. -Vol. 20. -No. suppl_1. -P. i15-i22. -URL:https://doi.org/10.1093/bioinformatics/bth918
Tran N. Knowledge-based framework for hypothesis formation in biochemical networks / Tran N. et al. //Bioinformatics. -2005. -Vol. 21 -No. suppl_2. P. ii213-ii219. -URL:https://doi.org/10.1093/bioinformatics/bti1134
Muggleton S.H. Machine Learning for Systems Biology / Muggleton S.H. //Inductive Logic Programming, Lecture Notes in Computer Science: Edited by Kramer S., Pfahringer B., Springer. -2005. -Vol. 3625. -P. 416-423. -URL:https://doi.org/10.1007/11536314_27
Tamaddoni-Nezhad A. Application of abductive ILP to learning metabolic network inhibition from temporal data / Tamaddoni-Nezhad A. et al. //Machine Learning. -2006. Vol. 64. -P. 209–230. -URL:https://doi.org/10.1007/s10994-006-8988-x
Muggleton S. Knowledge Mining Biological Network Models / Muggleton S. //6th IFIP TC 12 International Conference on Intelligent Information Processing (IIP). -2010. -P. 2-2. -URL:https://doi.org/10.1007/978-3-642-16327-2_2
Linde J. Data- and knowledge-based modeling of gene regulatory networks: an update / Linde J. et al. //EXCLI J. -2015. -Vol. 14. -P. 346-378. -URL:https://doi.org/10.17179/excli2015-168
Rougny A. A logic-based method to build signaling networks and propose experimental plans / Rougny A. et al.. //Sci Rep. -2018. -Vol. 8. -No. 7830. -URL:https://doi.org/10.1038/s41598-018-26006-2
Kitano H. Using process diagrams for the graphical representation of biological networks / Kitano H. et al. //Nat Biotechnol. -2005. -Vol. 23. -No. 8. -P. 961-966. -URL:https://doi.org/10.1038/nbt1111
Kitano H. Accelerating systems biology research and its real world deployment. //NPJ Syst Biol Appl. -2015. -Vol. 1. -No. 15009. -URL:https://doi.org/10.1038/npjsba.2015.9
Kitano H. Artificial Intelligence to Win the Nobel Prize and Beyond: Creating the Engine for Scientific Discovery / Kitano H. //AI Mag. -2016. -Vol. 37. -No. 1. -P. 39-49. -URL:https://doi.org/10.1609/aimag.v37i1.2642
Hsin K.Y. systemsDock: a web server for network pharmacology-based prediction and analysis / Hsin K.Y. et al. //Nucleic Acids Res. -2016. -Vol. 44. -No issue_W1. -P W507-W513. -URL:https://doi.org/10.1093/nar/gkw335
Emmert-Streib F., Dehmer M.,, Haibe-Kains B. Gene regulatory networks and their applications: understanding biological and medical problems in terms of networks / Emmert-Streib F., Dehmer M.,, Haibe-Kains B. //Front Cell Dev Biol. -2014. -Vol. 2. -No. 38. -URL:https://doi.org/10.3389/fcell.2014.00038
Demir E. The BioPAX community standard for pathway data sharing / Demir E. et al. //Nat Biotechnol. -2010. -Vol. 28. -No 9. P. 935–942. -URL: https://doi.org/10.1038/nbt.1666
Musen M.A. The Protégé Project: A Look Back and a Look Forward / Musen M.A et al. //AI Matters. -2015. -Vol. 1. -No. 4.. -URL:https://doi.org/10.1145/2757001.2757003
Fiorini N. Towards PubMed 2.0 / Fiorini N., Lipman D. J., Lu Z. //eLife. -2017. -Vol. 6. -No e28801. -URL:https://doi.org/10.7554/eLife.28801
Rose P.W. The RCSB protein data bank: integrative view of protein, gene and 3D structural information / Rose P.W. et al. //Nucleic Acids Res. -2017. -Vol. 45. -No. issue_D1. -P. D271-D281. -URL:https://doi.org/10.1093/nar/gkw1000
Berman H. M. The Protein Data Bank archive as an open data resource / Berman H. M. et al //J Comput Aided Mol Des. -2014. -Vol. 28. -No. 10. -P. 1009-1014. URL:https://doi.org/10.1007/s10822-014-9770-y
Gray K.A. A review of the new HGNC gene family resource / Gray K.A. et al. //Hum Genomics. -2016. -Vol. 10. -No. 6. -URL:https://doi.org/10.1186/s40246-016-0062-6
Thomas P.D. The Gene Ontology and the Meaning of Biological Function / Thomas P.D. //Methods Mol Biol. -2017. -Vol. 1446. -P. 15-24. -URL:https://doi.org/10.1007/978-1-4939-3743-1_2
Pundir S. UniProt Protein Knowledgebase / Pundir S, Martin MJ, O'Donovan C. //Methods Mol Biol. -2017. Vol. 1558. P. 41-55. -URL:https://doi.org/10.1007/978-1-4939-6783-4_2
Baumann N. How to use the medical subject headings (MeSH) / Baumann N. //Int J Clin Pract. -2016. -Vol. 70. -No. issue_2. -P. 171-174. -URL:https://doi.org/10.1111/ijcp.12767
Bhalla U. S. Understanding complex signaling networks through models and metaphors / Bhalla U. S. //Prog Biophys Mol Biol. -2003. -Vol. 81. -No. issue_1. -P. 45-65. -URL:https://doi.org/10.1016/s0079-6107(02)00046-9
Shao Li, Lijiang Wu, Zhongqi Zhang (2006). Constructing biological networks through combined literature mining and microarray analysis: a LMMA approach/ Shao Li, Lijiang Wu, Zhongqi Zhang. //Bioinformatics. -2006. -Vol. 22. No issue_17. -P. 2143–2150, https://doi.org/10.1093/bioinformatics/btl363
Li X. Global mapping of gene/protein interactions in PubMed abstracts: a framework and an experiment with P53 interactions / Li X. et al. //J Biomed Inform. -2007. -Vol. 40. -No issue_5. -P. 453-464. -URL:https://doi.org/10.1016/j.jbi.2007.01.001
Cho K.H. Reverse engineering of gene regulatory networks / Cho K.H. et al. //IET Syst Biol. -2007. -Vol. 1. -No 3. -P. 149-163. -URL:https://doi.org/10.1049/iet-syb:20060075
Camacho D. Comparison of reverse-engineering methods using an in silico network / Camacho D. et al. //Ann N Y Acad Sci. -2007. -Vol. 1115. -No. issue_1. -P. 73-89. URL:https://doi.org/10.1196/annals.1407.006
Karlebach G. Modelling and analysis of gene regulatory networks / Karlebach G., Shamir R. //Nat Rev Mol Cell Biol. -2008. -Vol. 9. -No 10. -P. 770-780. -URL:https://doi.org/10.1038/nrm2503
Viswanathan G.A. Getting started in biological pathway construction and analysis / Viswanathan G.A. et al. //PLoS computational biology. -2008. -Vol. 4. -No. 2. -P. e16. -URL:https://doi.org/10.1371/journal.pcbi.0040016
Janky R. iRegulon: from a gene list to a gene regulatory network using large motif and track collections / Janky R. et al. //PLoS Comput Biol. -2014. -Vol. 10. No. 7. -URL:https://doi.org/10.1371/journal.pcbi.1003731
Cussat-Blanc S. Artificial Gene Regulatory Networks-A Review / Cussat-Blanc S., Harrington K., Banzhaf W. //Artif Life. -2018. -Vol. 24. -No issue_4. -P. 296-328. -URL:https://doi.org/10.1162/artl_a_00267
Dávila J. Una gramática de estilos para resumir textos en español [A grammar of style to summarize Spanish texts] / Dávila J., Contreras H. //Revista de La Sociedad Español para el Procesamiento del Lenguaje Natural (SEPLN). -2002. -Vol. 29. -P. 265-273. -URL:http://www.saber.ula.ve/handle/123456789/15963 [in Spanish]
Parra M.M. Un Modelo Computacional para la Generación de Resúmenes Automáticos de Artículos Científicos en Español [A computational model for the generation of automatic, Spanish, scientific abstracts] / Parra M.M., Dávila J. //ACTAS del IX Simposio Internacional de Comunicación Social. -2005. -P. 213-216. -URL:https://www.researchgate.net/publication/44476598 [in Spanish]
Lu TT. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors / Lu TT. et al. //Mol Cell. -2000. -Vol. 6. -No 3. -P. 507-515. -URL:https://doi.org/10.1016/s1097-2765(00)00050-2
Schmidt O. (2009). A standard-compliant global map of the bile acid/xenobiotic signalling network: Construction and automated query processing / Schmidt O. et al. //Proceedings of The 2009 International Conference on Bioinformatics and Computational Biology (BIOCOMP'2009). -2009. -Vol. II. -P. 657-663. -URL:https://www.researchgate.net/publication/344755794
Lee J. Processing SPARQL queries with regular expressions in RDF databases / Lee J. et al. //BMC Bioinformatics. -2011. -Vol. 12. -No suppl_2. -P. S6. -URL:https://doi.org/10.1186/1471-2105-12-S2-S6
Ladunga I. Finding homologs to nucleotide sequences using network BLAST searches / Ladunga I. //Curr Protoc Bioinformatics. -2002. ; -Vol 00. -No. issue_1. -P. 3.3.1-3.3.25. -URL:https://doi.org/10.1002/0471250953.bi0303s00
Tsunoda T., Takagi T. Estimating transcription factor bindability on DNA / Tsunoda T., Takagi T. //Bioinformatics. -1999. -Vol. 15. -No. issue_7. -P. 622-630. -URL:https://doi.org/10.1093/bioinformatics/15.7.622
Wielemaker J. SWI-prolog / Wielemaker, J. et al. //Theory and Practice of Logic Programming. -2012. -Vol. 12. -No 1-2. -P. 67–96. -URL:https://doi.org/10.1017/S1471068411000494