Двойная деконволюция опухолей молочной железы у собак выявляет значительное обогащение CD4+ T-клеток с помощью EPIC и CIBERSORTx
Двойная деконволюция опухолей молочной железы у собак выявляет значительное обогащение CD4+ T-клеток с помощью EPIC и CIBERSORTx

Аннотация
Опухоли молочной железы собак (ОМС) являются отличной моделью рака молочной железы человека благодаря своему спонтанному происхождению и иммунологическому сходству. В данном исследовании проведена двойная иммунная деконволюция — EPIC и CIBERSORTx — данных транскриптомного секвенирования (GSE136197) из 48 образцов опухолевых и нормальных тканей молочной железы собак для определения иммунного микроокружения опухоли. В опухолевых образцах выявлено значительное и статистически достоверное обогащение CD4⁺ T-клеток (коэновское d = –0,83, FDR < 0,01), которое прослеживается уже на стадии карциномы in situ. Анализ CIBERSORTx дополнительно позволил выделить покоящиеся и активированные клетки памяти CD4⁺ T, а также T-фолликулярные хелперы, что подчеркивает функциональное разнообразие. Полученные результаты свидетельствуют о сохранённом механизме иммунного ремоделирования с участием CD4⁺ T-клеток у собак и человека и демонстрируют, что комбинированная деконволюция EPIC–CIBERSORTx является экономичным способом получения высокодетализированных иммунных профилей, способных служить основой для сравнительной онкологии и разработки иммунотерапии.
1. Introduction
Canine mammary tumors (CMTs) are the most common neoplasms in intact female dogs and share key characteristics with human breast cancer, including spontaneous tumor development, hormonal dependence, and similar age at onset and histological subtypes
. Molecular parallels include comparable expression patterns of estrogen and progesterone receptors, epidermal growth factor receptors, p53, Ki-67, and cyclooxygenases, supporting CMTs as useful models in comparative oncology . Prognostic markers such as lymph node involvement, tumor size, and clinical stage also align across species .The tumor immune microenvironment (TIME), composed of immune and stromal cells, plays a critical role in breast cancer progression and treatment response
. Subtypes like HER2-positive and triple-negative breast cancer show greater tumor-infiltrating lymphocytes (TILs), while luminal subtypes have lower immune infiltration and poorer outcomes . Specific immune cells, such as cytotoxic lymphocytes, regulatory T cells, mast cells, and tumor-associated macrophages, influence immune escape and therapeutic resistance . TIME subclassification frameworks (for example, inflamed, immune-desert, and immune-excluded) have been developed in human oncology to guide immunotherapy strategies [4], but their application to CMTs remains limited.Computational deconvolution of bulk RNA-seq enables estimation of immune cell fractions based on transcriptomic data, offering a scalable approach to TIME profiling
. EPIC (Estimating the Proportions of Immune and Cancer cells) and CIBERSORTx are such methods, validated across tissues and even across species, particularly for abundant immune populations like CD4⁺ and CD8⁺ T cells, B cells, macrophages, and cancer-associated fibroblasts . Benchmarking studies have shown that deconvolution tools like EPIC, CIBERSORT, and MCP-counter perform well with high-quality reference signatures, though limitations exist for under-characterized species or cell types .This study uses EPIC to profile immune infiltration in publicly available CMT transcriptomic data. The aim of the work is to determine whether CD4⁺ T cell abundance, a key immune correlate in human breast cancer, is also a distinguishing feature in canine tumors, thus contributing to cross-species immuno-oncology insights.
2. Research methods and principles
Sample collection and RNA sequencing:
This study re-analyzed raw RNA-seq data from 48 canine mammary gland samples (normal tissue, atypia-normal, carcinoma in situ, and invasive carcinoma) generated by Graim et al. (GSE136197). In the original work, tumor and matched normal tissues were obtained at surgical excision or diagnostic biopsy from privately owned dogs with informed owner consent and institutional ethical approval. Immediately after resection, tissues were flash-frozen in liquid nitrogen and stored at -80 °C until processing. Total RNA was extracted using TRIzol reagent, and integrity was confirmed by Bioanalyzer (RIN ≥ 7). Poly(A)-enriched, strand-specific cDNA libraries were prepared with the Illumina TruSeq RNA Sample Preparation Kit and sequenced on an Illumina HiSeq 2500 platform to a depth of ~50 million paired-end reads per sample.
Metadata curation and tumor quality control:
Sample-level metadata, including histological classification, versus normal status, and sequencing run information, were obtained from the original publication and the associated GEO records. All annotations were cross-checked and harmonized as follows: (i) sample identifiers were matched between the supplementary tables of Graim et al. and the GEO sample sheets; (ii) histological labels were standardized to four categories (normal, atypia-normal, carcinoma in situ, invasive carcinoma); (iii) any records with incomplete or ambiguous histology were excluded (none met this criterion); and (iv) sequencing metrics such as total read count and mapping rate were screened to confirm data completeness. The final curated metadata table was merged with immune cell fraction outputs for downstream statistical analysis. The data were then reshaped into long format using the pivot_longer function from the tidyr package, which facilitated efficient plotting and subgroup comparisons.
Data preprocessing for immune deconvolution:
Raw read counts were transformed into Transcripts Per Million (TPM) to standardize expression values across samples and ensure compatibility with the algorithms used. Because EPIC and CIBERSORTx were originally developed for human transcriptomes, Ensembl canine gene identifiers were mapped to their corresponding human orthologs using the : biomaRt package, retaining only one-to-one orthologs to preserve downstream cell-type annotation fidelity and avoid ambiguities from gene duplication or divergence.
Statistical analysis:
All statistical analyses were performed in R version 4.2.3 using the packages tidyverse, pheatmap, FSA, and effsize. Because immune cell fractions were not normally distributed, group-wise comparisons were carried out with the Kruskal–Wallis test, followed by Dunn's post hoc pairwise tests. P-values were adjusted for multiple testing using the Benjamini–Hochberg false discovery rate (FDR) procedure, and statistical significance was defined as FDR-adjusted p < 0,05. Effect sizes were calculated with Cohen's d and interpreted as small (≥ 0,2), medium (≥ 0,5), or large (≥ 0,8). All figures and heatmaps were generated directly from these R workflows to ensure full reproducibility.
Immune cell proportions were estimated using two computational deconvolution tools: EPIC (Estimating the Proportions of Immune and Cancer cells) and CIBERSORTx. The EPIC R package was applied using its default reference signature, which estimates the abundance of major immune and stromal cell types, including CD4⁺ T cells, CD8⁺ T cells, B cells, natural killer (NK) cells, macrophages, cancer-associated fibroblasts (CAFs), and endothelial cells. The category “otherCells”, which includes uncharacterised stromal or malignant cells, was also retained to provide a comprehensive overview of cellular heterogeneity. In parallel, CIBERSORTx was used via its web-based interface, leveraging the LM22 signature matrix that defines 22 human hematopoietic cell types. The TPM-normalised, human ortholog-converted matrix was uploaded without batch correction, as all samples originated from a single dataset. CIBERSORTx was run in relative mode, and immune cell fraction estimates were downloaded for integration with EPIC results.
To visualize immune infiltration patterns, z-score scaled heatmaps were generated using the pheatmap package, with scaling applied across rows to highlight differences in immune cell abundance between histological subtypes. Boxplots were created using ggplot2 to compare the distributions of specific immune cell populations, particularly CD4⁺ T cells, across different histological groups. To quantify the magnitude of immune differences, effect sizes were calculated using the cohen.d function from the effsize package, with thresholds for interpretation set at 0,2 (small), 0,5 (medium), and 0,8 (large). Particular attention was given to CD4⁺ T cells due to their established relevance in human breast cancer immunology.
Given the heterogeneous distribution and small sample sizes within each subgroup, non-parametric statistical testing was used throughout. Kruskal–Wallis tests were employed for overall comparisons between histology groups, followed by Dunn's post hoc tests using the dunnTest function from the FSA package to identify pairwise differences. To control the false discovery rate, all p-values were adjusted using the Benjamini–Hochberg procedure. All code and analytical workflows were implemented in Jupyter Lab with an R kernel and are available upon request to ensure reproducibility.
Data availability:
The full analysis pipeline, including code scripts for EPIC deconvolution, ortholog mapping, and figure generation, is available at the following GitHub repository: https://github.com/ruwinimadhu/Immune-Deconvolution-GSE136197. The processed data derived from GSE136197 and human ortholog mappings are included in the repository for reproducibility.
The data used for EPIC analysis were downloaded from the NCBI Gene Expression Omnibus (GEO) under accession number GSE136197
. The dataset is available under a CC BY-NC 4.0 license.Funding & ethics disclosure:
This research did not receive any specific grant from funding agencies. No new animal experiments were conducted. The analysis was performed on publicly available data.
3. Main results
Computational deconvolution of RNA-seq data from 48 canine mammary tissue samples using EPIC revealed a distinct reshaping of the tumor immune microenvironment (TIME) between tumor and normal samples. A consistent increase in CD4⁺ T cell infiltration was observed across all histological subtypes in tumor tissues. This increase was statistically significant (Cohen's d = -0,83, p < 0,01), representing a large effect size and indicating a biologically meaningful elevation in helper T cell recruitment or expansion (Fig. 1). Although CD8⁺ T cells showed a modest upward trend, differences in macrophages, B cells, and NK cells were less pronounced (Cohen's d < 0,5) and did not reach statistical significance.
These findings were visually reinforced by heatmap clustering of row-wise z-scoresTo further dissec, which demonstrated clear separation between tumor and normal samples, primarily driven by differences in CD4⁺ and CD8⁺ T cell levels. Statistical comparisons using the Kruskal–Wallis test confirmed that CD4⁺ T cell proportions varied significantly across histological groups (p < 0,01), and Dunn's post hoc analysis pinpointed significant differences between carcinoma and normal tissue. Notably, CD4⁺ T cell enrichment was already evident in early lesions, such as carcinoma in situ, suggesting that immune remodeling is an early event in canine mammary tumor progression.
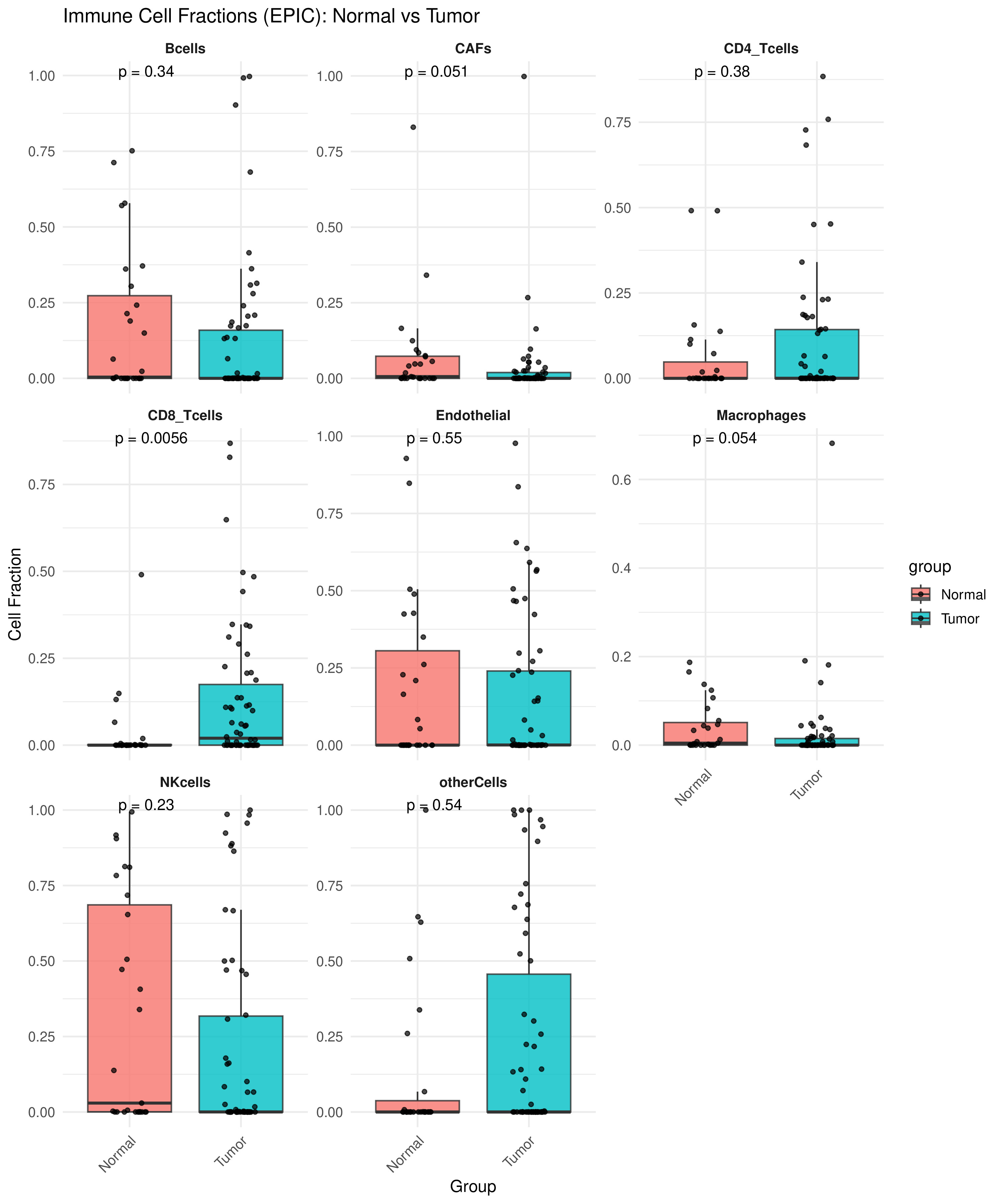
Immune cell fraction in normal vs tumour tissues estimated using EPIC deconvolution
several immune cell types showed a significant difference between groups. Notably, CD4+ T cells were significantly enriched in tumour tissues
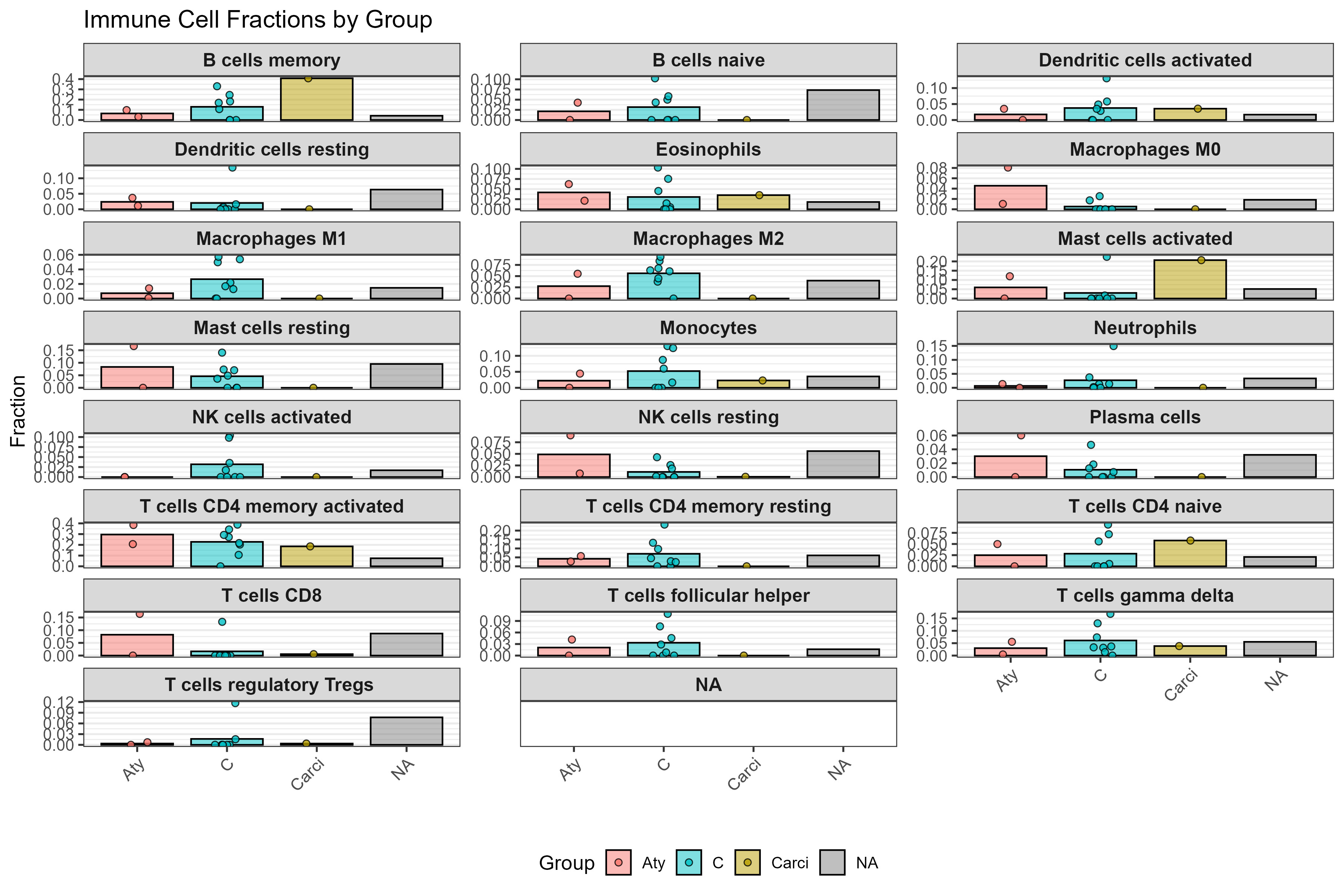
CD4⁺ T cell subset composition across histological subtypes of canine mammary tumours as estimated by CIBERSORTx
boxplots show relative fractions of CD4⁺ memory resting T cells, CD4⁺ memory activated T cells, Tregs, and T follicular helper cells, highlighting functional diversity within the CD4⁺ compartment across tumour progression stages. These patterns reflect early immune activation and possible immune escape mechanisms
4. Discussion
While
used the same GSE136197 cohort to model the molecular development and oncogenic drivers of canine mammary tumors, their focus remained on transcriptional trajectories and cancer‐initiating pathways. In contrast, the present work interrogates the tumor immune microenvironment by applying a dual deconvolution strategy (EPIC and CIBERSORTx) to quantify CD4⁺ T-cell enrichment and to resolve key CD4⁺ subsets. This immune-centric analysis provides a higher-resolution view of lymphocyte composition and positions CD4⁺ T cells as candidate biomarkers and therapeutic targets, extending the dataset’s translational value toward comparative immuno-oncology and the design of future immunotherapy studies.The elevation of CD4⁺ T cells observed in this study aligns with findings in human breast cancer, where altered CD4⁺ T cell infiltration correlates with immune evasion, therapy response, and tumour subtype
, . CD4⁺ T cells play diverse roles in coordinating adaptive immunity via cytokine signaling, antigen presentation, and effector cell recruitment . Their infiltration is often associated with both prognostic value and therapeutic responsiveness in human disease.The identification of resting and activated memory CD4⁺ T cells suggests a combination of immune surveillance and active response states. T follicular helper cells may indicate the formation of tertiary lymphoid structures, further supporting immune involvement in the tumour microenvironment. Although Tregs were infrequent, their presence hints at early immunosuppressive activity, potentially contributing to tumour immune escape. This functional heterogeneity of CD4⁺ subsets reflects similar trends seen in human breast cancer, where the balance between effector and regulatory cells can shape disease progression and treatment outcomes.
By enabling this level of resolution, CIBERSORTx complements EPIC and strengthens the translational utility of immune deconvolution in comparative oncology. The conserved immune patterns between canine and human mammary tumours reinforce the value of canine models for immunotherapy research.
However, limitations must be acknowledged. Both EPIC and CIBERSORTx rely on human-derived reference signatures, which may introduce bias when applied to canine data. Moreover, they lack spatial context and cannot capture dynamic functional states such as T cell exhaustion. The absence of clinical metadata in this study also limits the ability to link immune features with prognosis or treatment outcomes.
Despite these limitations, this study demonstrates the feasibility and utility of applying dual-method immune deconvolution pipelines to veterinary cancer data. The consistent elevation of memory and activated CD4⁺ T cell subsets suggests their potential as biomarkers of tumour immunity and as targets for future therapeutic strategies in canine mammary tumours.
5. Conclusion
This study demonstrates a significant increase in CD4⁺ T cell infiltration within the tumour immune microenvironment of canine mammary tumours (CMTs), as revealed by EPIC deconvolution of RNA-seq data. The consistent enrichment of CD4⁺ T cells across histological subtypes — including early lesions such as carcinoma in situ—suggests that immune remodelling is an early and sustained feature of CMT progression. These findings position CD4⁺ T cells as potential biomarkers and therapeutic targets in veterinary oncology, while also underscoring key immunological parallels between canine and human breast cancer. The use of computational deconvolution tools such as EPIC and CIBERSORTx highlights the feasibility of cost-effective immune profiling using existing transcriptomic datasets. To advance the clinical relevance of these findings, future studies should incorporate clinical outcome data, spatial immune profiling, and single-cell analyses to further elucidate the functional and prognostic roles of immune cells in CMTs.