Конвейер обработки данных с использованием искусственного интеллекта для моделирования и анализа сетей регуляции генов
Конвейер обработки данных с использованием искусственного интеллекта для моделирования и анализа сетей регуляции генов

Аннотация
Этот проект объединяет генеративный ИИ и логический ИИ для создания интегрированного процесса, конвейера, который извлекает большие объемы научной информации и создает логические модели, которые могут быть проверены и использованы экспертами. Наши цели — организовать и проанализировать с помощью ИИ и других инструментов поиска биоинформации документацию, связанную с определенной областью, и извлечь проверенные знания для поддержки планирования решения проблем. Мы протестировали нашу методологию и конвейер на эксперименте с COVID-19, о котором также сообщаем. Инструмент помог команде биологов оценить современное состояние дел и справиться с резким ростом числа научных работ во время пандемии. Основным продуктом конвейера является сеть генной регуляции (GRN); семантическая карта, представляющая собой отображение причинно-следственных связей в системной биологии, которая, в свою очередь, может быть использована для изучения различных стратегий вмешательства и лечения. Конвейер (названный Biopatternsg) был структурирован вокруг трех основных задач: а) сбор данных и информации; b) использование генеративного ИИ и больших языковых моделей (LLM) для автоматического извлечения биологических объектов и их биологических взаимосвязей, и c) использование логического ИИ для управления информацией, полученной в пунктах a) и b), и ее объединения с консервативными критериями, представленными в виде баз знаний на языке Prolog. Система также отслеживает и обеспечивает доступ ко всем исходным документам, используемым в качестве источников знаний. Мы считаем, что этот конвейер полезен для углубления понимания COVID-19. Мы также организовали систематическую оценку biopatternsg как метода для создания генных регуляторных сетей (GRN). Оценка распознавания имен показывает средний показатель F1-Score 0,9069 с дисперсией 0,0145 среди 30 сетей, используемых в качестве эталонного стандарта. Это указывает на отличную способность распознавать названия реальных биологических объектов. Результаты на других уровнях оценки не очень хороши, например, средний показатель F1-Score составляет 0,2306 с дисперсией 0,0218. Стоит отметить значительную разницу между точностью (Precision) и полнотой (Recall). Система даже демонстрирует идеальную полноту (1,0) в некоторых сетях, но точность всегда ниже 0,5, что снижает показатель F1-меры. Мы обсуждаем эти результаты и объясняем, почему даже частичный успех с полнотой является обнадеживающим.
Репозиторий Biopatternsg находится по адресу: https://github.com/biopatternsg/biopatternsg
Дополнительные материалы находятся в: https://github.com/biopatternsg/biopatternsg/tree/feature/evaluation
1. Introduction
In his 2011 book, "Thinking, Fast and Slow," Nobel Prize winner Daniel Kahneman
, explains that human thinking could be seen as having two components: System 1, which is fast and intuitive, and System 2, which is slower and responsible for explainable reasoning. The same phenomenon is observed in Artificial Intelligence: Generative AI is fast and intuitive and can be used to identify patterns in huge volumes of data. Logical AI enables systematic, verifiable, and explainable reasoning on this preprocessed data. This project combines both types of AI to form an integrated process, a pipeline, that retrieves large volumes of scientific information and produces logical models that can be validated and leveraged by human experts.We propose a connection between generative AI and logic AI to define a framework for knowledge modeling, management, and discovery that enables the automatic construction of knowledge networks for a domain. The main idea here is to retrieve and analyze, with the help of AI, the documentation associated with a domain and extract from it the elements and possible relationships that connect them, thus facilitating its understanding and assisting in the planning of actions for problem solving (See Figure 1).
Figure 1 depicts the general components in our proposal as a pipeline that connects three main tasks:
a) access to various sources for information gathering inherent to a domain and problem of interest;
b) use of generative artificial intelligence to assist in the automatic extraction of knowledge;
c) use of logic artificial intelligence to automatically organize and explore knowledge bases, following constraints that guide such exploration.
The final result in our work are models (graphs or networks) that synthesize knowledge in such a way as to facilitate decision-making related to problem solving. And it is our main goal to answer queries like: given a drug, what kinds of protein-protein interactions result when that drug binds to a known receptor, and which of them lead to the activation (or inhibition) of the transcriptional response of some gene?. We also consider questions such as: for a given pair of proteins, is there a subnet in this modeled GRN that describes interconnected regulatory pathways, in which such proteins stimulate and inhibit their mutual transcription in any way? .
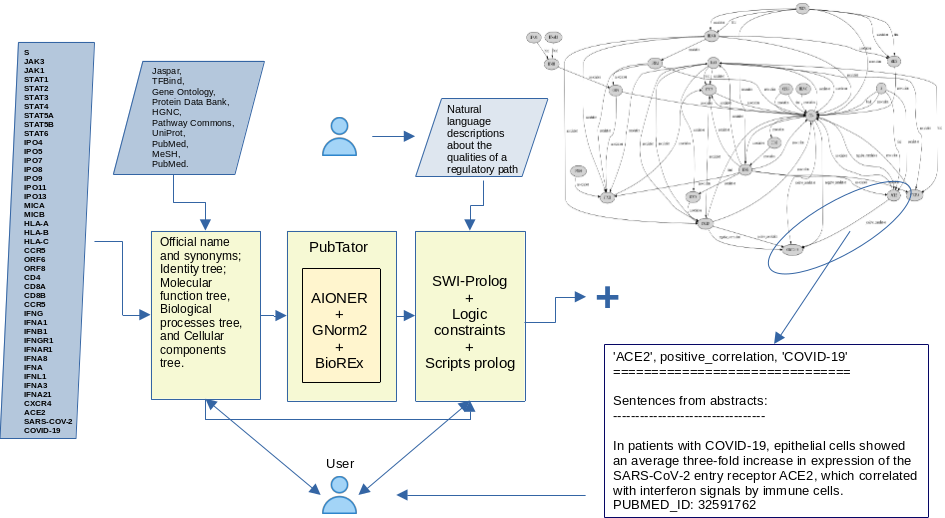
A schematic view of the modeling and type of results for COVID-19
1.1 Gene Regulatory Networks (GRNs)
Gene transcription regulatory networks (GRNs) are biological mechanisms responsible for regulating the presence or absence of gene-associated products. In a GRNs, a regulatory event can activate a product that in turn participates in an event, which activates or inhibits another product. It is normal, then, that in such networks the complexity of the interrelationships grows very rapidly , .
In order to manage the current knowledge inherent to GRNs, informatics strategies have been developed aimed at the description, organization, interrelation and analysis of the elements that constitute them. Among such strategies are ontologies , , , and process diagrams , .
Here, we are interested in the modeling and analysis of Gene transcription regulatory networks, based on the inferential processing of knowledge bases ; therefore, our work fits in the ontology-based approach and in their logical and analysis facilities. Elaborating on that, we have proposed the automatic construction and integration of artificial intelligence and various knowledge bases (KBs), in order to take advantage of the great effort that the scientific community has made about GRNs, regarding their organization and availability.
1.2 The Pipeline
Figure 1 depicts the flow of information in our system applied to the COVID-19 disease. In this scenario, a biologist or physician explores possible treatment alternatives for the COVID-19, providing some inputs and obtaining a semantic graph that represents a GRN, enriched with a set of knowledge bases and detailed documentation about the regulatory events and pathways that shape that network. Below are some details of the pipeline’s inputs, components, and results:
1. Initial list of descriptors (concepts, keywords) provided by the user. For this particular domain, the descriptors are biological or chemical objects; for instance: drugs, small molecules, genes/proteins of various types, and the name of the disease to analyze.
2. Bioinformatics portals accessible through the internet, which describe objects such as those mentioned in the previous item; for instance MeSH, Gene Ontology and PubMed (pubmed.ncbi.nlm.nih.gov), a portal specialized in information related to biology and medicine.
3. Generative artificial intelligence, based on Large Language Models (LLMs), specially trained to handle the information available in PubMed. It allows for:
a) the automatic recognition of biological entities related to COVID-19 (NER Model: Named Entity Recognition);
b) the automatic extraction of the biological relationships that connect them (RE Model: Relation Extraction).
The output obtained from the LLMs consists of entities and relationships that allow the construction of a knowledge base, useful for automatic reasoning with symbolic AI.4. An inference system based on symbolic AI, used for automatic reasoning processes on the knowledge bases organized with the assistance of LLMs, and repositories, like the ones visible in Figure 2, Figure 3 and Figure 4. The user explains the desirable characteristics for the solutions (paths) to be obtained to guide the logical inference system in the deduction of possible paths to solutions. The output in this scenario is a GRN like the one shown in Figure 5. For a detailed view of Figure 5 see VERY-RESTRICTED/covid-19-wide-restricted-list-no-sm.png in supplemental material, in which the user can see how biological objects interact with each other, and how these interactions could eventually lead to the inhibition of COVID-19.
4. An inference system based on symbolic AI, used for automatic reasoning processes on knowledge bases organized with the assistance of LLMs and repositories, like those shown in Figure 2 and Figure 3. The user explains the desirable characteristics for the solutions (paths) to be obtained to guide the logical inference system in deducing possible paths to solutions. The output in this scenario is a GRN like the one shown in Figure 5 (see covid-19-wide-restricted-list-no-sm.svg for a detailed view), in which the user can see how biological objects interact with each other, and how these interactions could eventually lead to the inhibition of COVID-19.
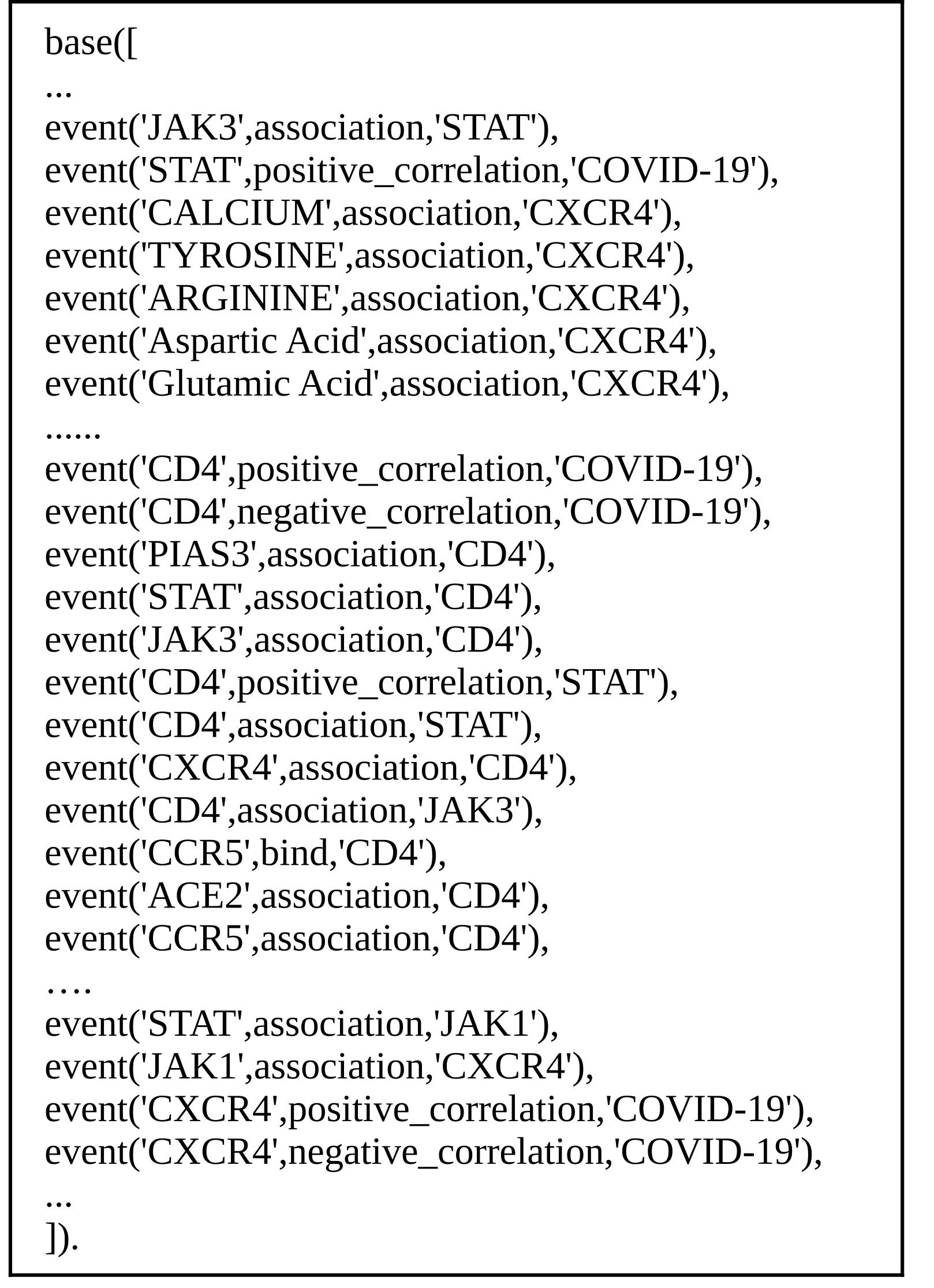
Regulatory events knowledge base (KB)
the KB was modeled automatically and contains more than 20,000 events

KBs organized following the steps in Fig. 1 and Fig. 6 (Part 1)

KBs organized following the steps in Fig. 1 and Fig. 6 (Part 2)

An inferred pathway correlating ACE2 and COVID-19
2. The Workflow to Build the Knowledge Bases
Subnetworks for the regulation of COVID-19 and CXCR4 (Part 1)
1) names, synonyms, biological objects, and related scientific documents from the MeSH service; Gene Ontology ; PubMed , Protein Data Bank (PDB) , , HGNC (HUGO Gene Nomenclature Committee) , and UniProt ;
2) object identity, based on the the MeSH service;
3) molecular function, biological processes, and cellular components associated with each network object, from Gene Ontology;
4) definitions, which can be extracted via automatic inferences performed from 1, 2, and 3, establishing that an object satisfies the constraints guiding the analysis of pathways and subnetworks.
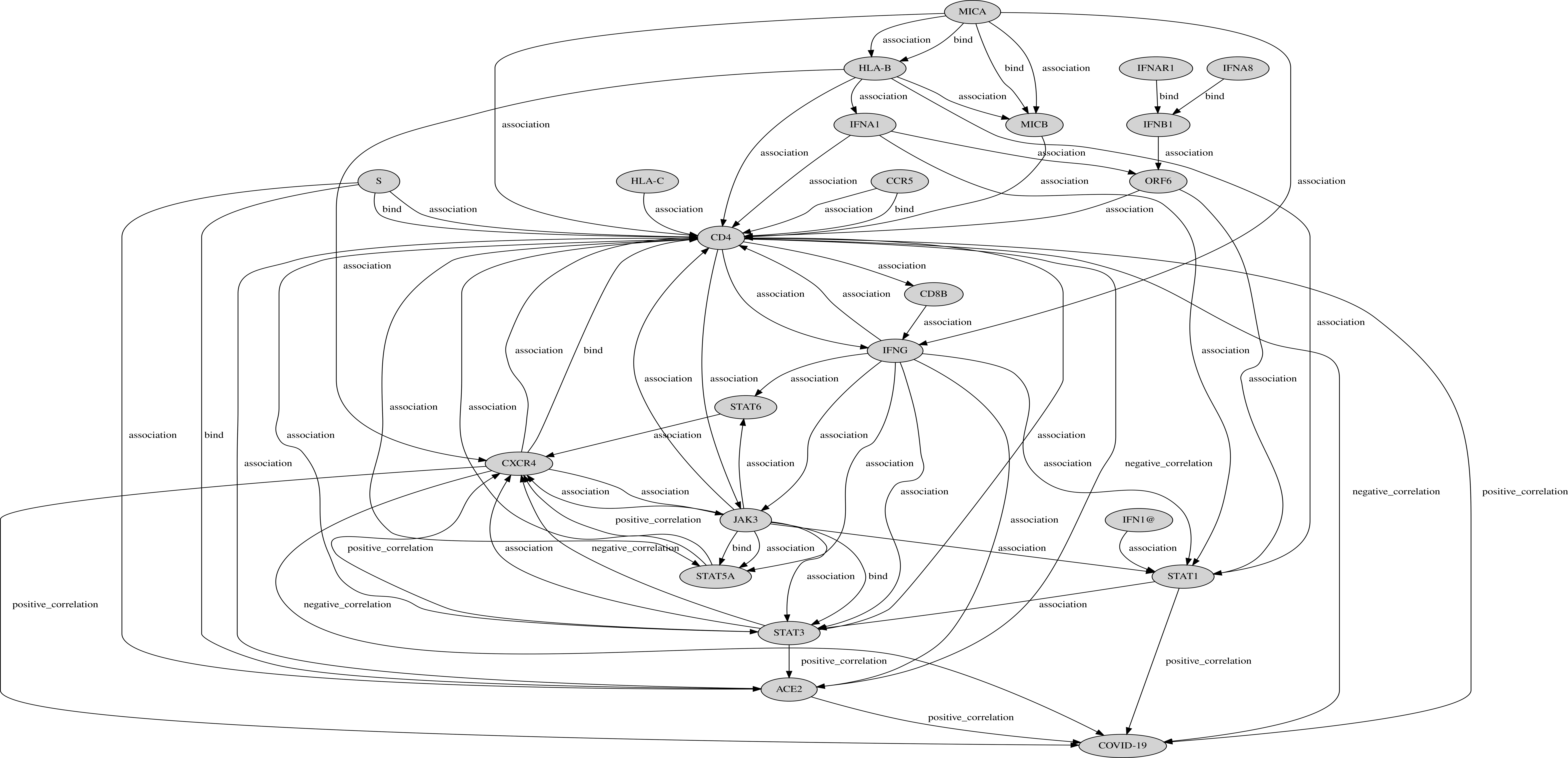
A restricted GRN for the exploration of regulatory subnetworks for COVID-19 and CXCR4
Subnetworks for the regulation of COVID-19 and CXCR4 (Part 2)
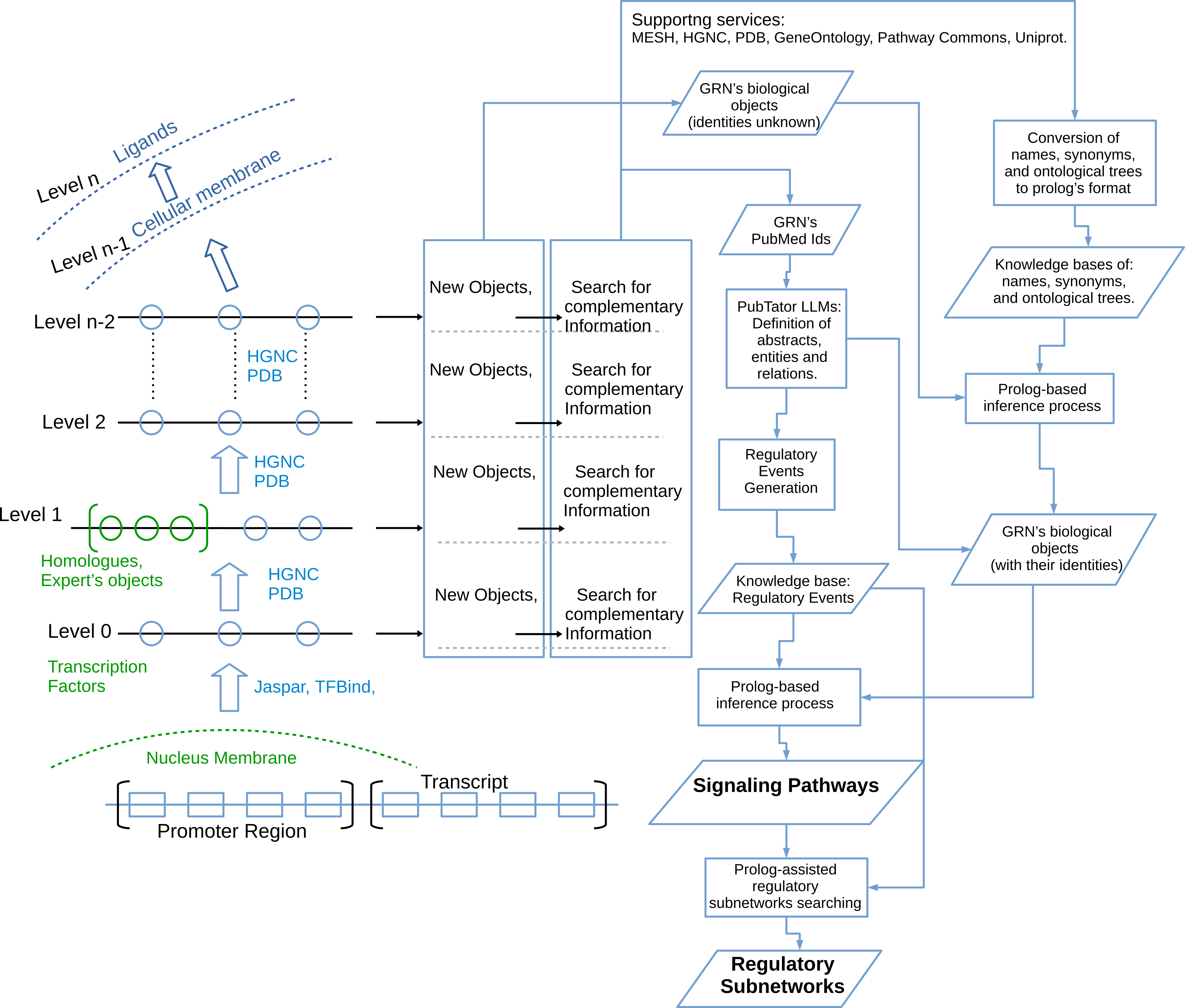
Work-flow for the semantic modeling and analyzing of a GRN
1. Definition of potential transcription factors linked to the regulatory region of specific proteins of interest, based on TFBIND and JASPAR .
2. Search for potential biological objects (e.g., ligands and proteins) linked to both the system-proposed transcription factors and other biological objects provided by the user, using the Protein Data Bank , and UniProt.
3. Generation of knowledge bases describing the identity and functionality of network objects (including ligands, proteins, enzymes, and receptors), based on HGNC , UniProt, MeSH , and Gene Ontology .
4. Generation of keyword combinations among related objects in the network, considering not only the names defined by the modeler but also any synonyms defined for them.
5. Retrieval of PubMed IDs to subsequently obtain abstracts addressing the objects defined for the network, via PubMed .
6. Automatic generation of a corpus of abstracts and annotations using the NCBI PubTator service , utilizing the PubMed IDs defined in the previous step.
7. Automatic extraction of regulatory events associated with the Gene Regulatory Network (GRN) objects from the corpus of abstracts and annotations collected from PubTator. Generation of Prolog KBs.
8. Inference processes on Prolog code to discover regulatory pathways and subnetworks from the KBs modeled for the network.
Once the modeling levels depicted in Figure 7 are complete, we searched PubMed for a set of abstracts related to each possible interaction between the objects in the initial GRN. To do this, the user-provided objects were organized into pairs, and PubMed was asked for a specified number of abstracts for each pair. In this case, we used a user-configurable threshold of 200 abstracts, as this number is usually sufficient to cover the most relevant and recent publications on each interaction in recent years. However, we could repeat the modeling process with a different threshold to obtain more information. Then, we use the services provided by the NCBI PubTator service, to download the abstracts, accompanied by annotations, which include the recognition of the biological objects and the extraction of their relations. PubTator identifies all the objects in the abstracts, which normally includes objects not defined in the previous steps of the modeling process.
2.1 About Generative AI in our pipeline
There is a huge amount of biological and medical information and knowledge, available today through different bioinformatics resources (some of them mentioned above). On the other hand, other resources have recently emerged, based on Generative Artificial Intelligence (GenAI) and Large Language Models, LLMs, to extract biological entities and their relationships from any publication available in portals such as PubMed . In this paper we use one of them, specifically PubTator , and explore how independent knowledge representations, associated with the different resources mentioned above, can assist and complement each other. In other words, we show how the entities and relations extracted from publications, using a collection of LLMs, can be connected to other KBs to facilitate the analyzing process of the knowledge gathered and modeled. The NCBI’s PubTator service relies on a set of LLMs to implement a pipeline with three main tasks:
a) biological entities recognition, implemented with an LLM named AIONER ;
b) a set of LLMs and tools, to normalize the names of biological entities, implemented as a tool named GNorm2 ;
c) an LLM dedicated to the task of relation extraction from PubMed abstracts, implemented through the BioREx LLM .
2.2. Modeling COVID-19 Knowledge Domain
Figure 1 shows that the user participates directly in the first and last pipeline’s stages. On the first stage, for instance, the user has access to a KB that she can check and improve the names and synonyms for each object. Those names and synonyms are critical later on when the pipeline defines the PubMed IDs, that the generative AI uses to assist the construction of the KB in Figure 2. On the other hand, at the pipeline’s last stage, the user has access to all the prolog KBs that the system automatically produces. For instance, the user can add or modify the identity descriptors of an object; the system can infer (using prolog) that an object is just a protein, but the user could improve the description by writing down that the object is a receptor and an enzyme too. This kind of human intervention has an effect on the quality of the pipeline’s results. The human-in-the-loop approach is a fundamental feature in our proposal; and we reinforced such quality, by offering the user the facility of re-executing the pipeline, from some point onwards, once she has modified a particular KB.
3. Inferring Regulatory Pathways and Regulatory Subnetworks
In our work, a set of constraints define the characteristics of the objects that will shape regulatory pathways. For instance, the regulatory pathways could end with certain types of transcription factors, or to begin with certain types of drugs, or to include only objects with specific molecular functions and biological processes. Using the constraints, the system can explore the knowledge bases described in Figure 3 and Figure 4, searching for the objects that satisfy the desired characteristics, and then, shape the objects’ definitions that will be used later when inferring the pathways (see section 4, Figure 4). In this scenario, a regulatory pathway is a causal chain of regulatory events such that their biological objects satisfy certain kinds of constraints regarding their biological identities, molecular functions, biological processes, and the ways they are related (bind, stimulation, inhibition, and so on).
Our system keeps track of and allows access to all the original documents used as sources of knowledge. It is possible, for instance, to choose an inferred regulatory pathway and retrieve the abstracts that support it (see Figure 5). A GRN, in our work, is the collection of regulatory pathways that it is possible to infer from the KBs modeled, and that fulfill the set of user’s constraints for the biology system (and problem) on consideration. Figure 6 is an instance of a GRN that satisfies the description formulated before. In the pathways shown in Figure 6, each pathway begins either with a ligand, receptor, or transcription factor, that binds to or associates with a protein. Subsequently, a chain of protein interactions shapes the pathways until an ending regulatory event is inferred, in which a receptor, or transcription factor, interacts either to an ending protein (ACE2, CXCR4) or to the COVID-19 disease. In Biopatterngs's pipeline the logic AI stage offers ways to configure the set of constraints that guide the way the pathways are searched.
Figure 6 shows that there is no interest (at that moment) on small molecules or drugs working in the network; but that can be easily changed by incorporating them and their roles at inference time (see RESTRICTED/covid-19-restricted-list-and-small-molecules-from-pubtator.png in supplemental material). Figure 6 also shows that the objects visible in it are limited to the list of the proteins initially delivered to the system (see Figure 1). However, this can also be configured to allow the incorporation during inference time of other gene/gene products, small molecules, or diseases, provided by the generative AI stage of the pipeline (instances of these in RESTRICTED/covid-19-genes-from-pubtator.png and RESTRICTED/covid-19-diseases-from-pubtator.png in supplemental material).
Figure 6 allows one to have a first look at biological interactions and possible pathways, and then choose some of particular interest. Suppose that we choose from Figure 6 the following set of objects: the Spike (S) protein and CD4 receptor, the interferon protein (IFNG), the receptors STAT3, STAT5A and ACE2, the enzyme JAK3 and, of course, COVID-19. We can also see in Figure 6 a negative correlation between the CXCR4 receptor and the CODID-19 disease. Therefore, we can decide to search for subnetworks showing CXCR4, mediating a shift in the regulatory process of the disease. Then, the following question can be formulated: does the regulatory network in Figure 6, includes any subnetworks branching off from the regulatory pathway initially chosen (the one in Figure 5), such that the COVID-19 regulation shifts from up-regulated to down-regulated, mediated by the receptor CXCR4?. The process to answer the question above goes as follows:
1. The searching process is restricted to a list of objects: Spike (S), CD4, IFNG, STAT3, STAT5A, ACE2, JAK3, COVID-19, and CXCR4; therefore, we ask to the Biopatternsg system to restrict the knowledge base of regulatory events to those objects, and then to execute the inference of possible pathways related with them. This step produces the new network depicted in Figure 8 (for a detailed view see VERY-RESTRICTED/subnetworks/covid-19-CXCR4-wide-subnetwork.png in supplemental material).
2. We provide to the system the pathway that must guide the searching of regulatory subnetworks for COVID-19 (the one in Figure 5).
3. Biopatternsg consults the knowledge base of regulatory events, looking for events that might serve as regulatory links, between the initial pathway and possible others that could change the regulatory state of the disease.
4. Once the system has produced a collection of candidate subnetworks and we make a revision of them, a three pathways subnetwork is selected for COVID-19; for instance, the one described in Table 1 (a and b) and Table 2 (c) .
5. Normally we want to know a way to regulate the protein that mediates the regulatory shifting in the initial regulatory process. Therefore, we run another search for a regulatory subnetwork for CXCR4, and after a revision, we select the subnetwork described in Table 1 (b) and Table 2 (d). Note that the second pathway mediating the regulation shift for COVID-19 visible in Table 1 (b), is used as the pathway that guides the searching for a regulatory subnetwork for CXCR4.
6. Finally, the subnetworks shown in Table 1 and Table 2 are documented using the documentation that the system provides for the knowledge base of regulatory events. Note that several sentences can explain a regulatory event and that not all of them correctly model the related event, but normally one of them does.
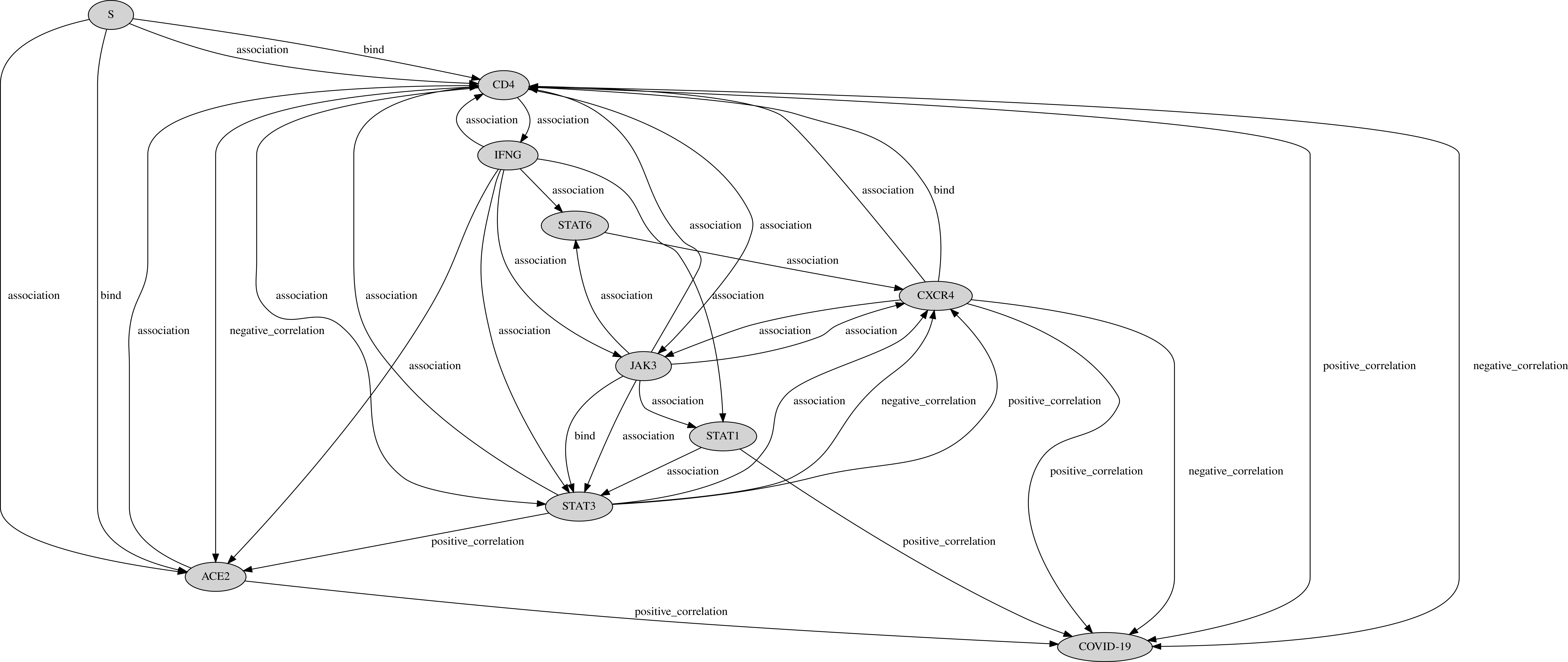
Restricted GRN to explore subnetworks for COVID-19 and CXCR4
Biopatternsg also offer resources to improve the list of objects initially provided as input to the system. For instance, once the first two stages of the Biopatternsg’s pipeline has finished, a report (named aligned.pl), is produced indicating which of the initially provided objects are already part of the knowledge base of regulatory events (KBase.pl), and which ones of them could be part of it, but represented using synonyms. Another report is also available (named synonyms.pl), regarding the main name of an object in the knowledge base and its synonyms; that report is produced using the metadata that accompanies the NER predictions made by PubTator’s LLMs. The report aligned.pl also shows which of the initial objects are not initially part of the knowledge base, and using both reports, aligned.pl and synonyms.pl, and the documentation of the knowledge base (named kBaseDoc.txt), it is possible also to explore alternative names for them (see examples of the files mentioned above in EVALUATION/CREB-phosphorylation). The result of all this is a better list of objects aligned to the names that the generative AI actually uses, for the objects that the researcher provided initially. Once a better aligned list has been elaborated, by means of the knowledge of who models the GRN, and the resources that Biopatternsg provides, then a new inference process can be run and an improved set of pathways and subnetworks can be obtained. This defines an iterative process of modeling and analysing that eventually could lead to important findings (see the researcher’s guide in DOC and the Biopatternsg’s repository wiki for a deeper description of its functionalities).
4. A framework for the evaluation of the method to produce GRNs
We have organized a systematic evaluation of biopatternsg as a method to produce gene regulatory networks, GRN. By a GRN we exactly mean the collection of objects and events reported in kBase.pl and it is the one we use here to evaluate Biopatternsg with the standard analysis of precision and recall (the F1 measures ).
In order to achieve this goal we retrieved, from Pathways Commons , a set of knowledge bases that serve as golden references to compare with. Pathways Commons offers facilities to export pathways in formats like sif (simple interaction format) and that allows us to create knowledge bases as the one in Table 7. We have compared the golden standard, or reference, from Pathways Common with the output of our system in three levels:
Level 1
At the first level, we checked whether a set of biological objects of interest, named in the sif files of a given GRN from Pathways Commons, can be identified by the pipeline’s tools. The key comparison is whether each name from each network used as reference is recognised by the AIONER LLM from Pubtator, the tool used by the pipeline.
We define as true positive, TP, as those names that are recognized by the tool and, of course, false negative, FN, those that fail to be recognized. The tool also suggests, in some cases, synonyms for given names which can be later verified to correspond or not to the original name in the GRN. So, those synonyms are a pessimistic estimate of false positives, FP, when they end up not corresponding to the original name. The usual definitions of Precision (TP/TP+FP) and Recall (TP/TP+FN) then hold. Table 3 lists 30 GRN provided by Pathways Commons and summarizes the results that we obtain for them.
The evaluation reports an average F1-Score of 0.9069 with a variance of 0.0145 among the 30 networks. This indicates an excellent capacity to recognize names of real, biological objects. It is worth noticing, also, that this F1 score is a minimum, as it could be higher when synonyms offered by the pipeline can be confirmed to be real names too.
We want to call the attention upon the column to the left of Table 3, sm/obs, which indicates the fraction of the number of names for small molecules (column sm, in the middle) divided by the amount of all the names in the GRN (column objects) provided as input to the pipeline. Notice that, as this fraction gets smaller, the F1 Score increases. We are actually using that column as an ordering key for the rest of the information in Table 3, a strategy that will prove to be useful to explain the results below at the other levels.

Pathways Commons sif file of the reactome’s pathway named creb phosphorylation (prolog format)
At the second level, the system is provided with pairs (object1, object2) as references, indicating relations in the GRN without actually naming the relations (a process confirmed to be noisy, as shown below).
We selected the referential GRN from Pathways Commons, of any size and with and without small molecules, to test PubTator’s NER and Normalization functions in those conditions. The procedure to build a knowledge base of regulatory events for each network/pathway in our experiments is as follows:
1) the sif (for comparison later) and the extended version of the sif file are downloaded from Pathways Commons;
2) using the extended sif file we collect the objects interacting in a network and, using the pipeline of Biopatternsg, a knowledge base of regulatory events is modeled for it;
3) the network's sif file is compared with the corresponding knowledge base produced by Biopatternsg. Step (3) is automated by a Prolog script available from the repo (see EVALUATION/README.txt and graph-comparison.pl in supplemental material). The data used for the comparison is also available in the repository (see EVALUATION).
Evaluation of names recognition in biopatternsg (level 1)
The results in Table 4 are discussed in the following section. The Average F1-Score is 0.3041 with a variance of 0.04 (Table 4 also reports an alternative F1-Score of 0.3652 obtained by adding up all TP, FP and FN from the 30 networks). The discrepancy may be due to the fact that the networks are intrinsically different and cannot be considered as one. For the CREB phosphorylation pathway, the aligned.pl file (see EVALUATION/CREB-phosphorylation) shows that all the objects visible in the reference are present in the knowledge base. It is a case of perfect recall.
Table 4 and Table 5 show the size of the knowledge bases for both reference and model, highlighting their high variability. The tables mentioned above highlight the cases where the reference size is larger and vice versa, and we can see that the cases are quite balanced.
Evaluation of pairs-like relations generation in biopatternsg (level 2)

An instance of report.txt for CREB-phosphorylation
At level 3 of the evaluation, we combine all the information produced by the pipeline to describe the GRN. At the level, the evaluation checks whether a triple (object 1, relation, object 2) appears in the reference. Unfortunately, the naming of relations differs between the golden standard and our system, so that we have to appeal to a series of synonyms, also produced by our system. Therefore, on the pipeline’s output we have something like (object 1, another name for the relation, object 2).
In any case, if a triple also appears in our output, it is counted as a true positive (TP). If the triple appears in the reference, but not in our output, is a false negative (FN). And if it does not appear in the reference, but our system produces it, it is a false positive (FP). Precision and Recall are calculated accordingly and as before. Table 5 lists the 30 GRN provided by Pathways Commons and summarizes the results of extracting the relation between pairs of biological objects.
The results overall are worse than at level 2. The F1-Score is 0.2306 with a variance of 0.0218, sensibly lower than in Table 4. The system exhibits perfect Recall (at 1) at more networks (4 in Table 5 vs. 1 in Table 4), but Precision drops even more and the distance between the two measures increases (and that reflects in that lower F1 Score).
We discuss these results in the following section and explain why even that partial success with Recall is an encouraging result.
Evaluation of relations extraction in biopatternsg (level 3)
5. Results and Discussion
Tables 4 and 5 show low averages for both the metrics (precision and recall). It is clear that, under the operational constraint of 200 abstracts retrieved for the model, the predictive capacity of Biopatternsg for relations is, on average, very limited. Precision is very low and gets worse when the name of the relation is actually compared (at level 3). However, Recall is perfect in more cases and increases between level 2 and level 3. In fact, in 10 out of 30 cases is a good Recall (above .70).
Low precision, however, requires special attention. The pipeline produces too many instances of events/relations that are not explicitly listed in the reference. A first hypothesis is that the reason for this may be the LLM (BioRex) behind PubTator has access to a wealth of documental references considerably bigger than the one used to sustain the GRNs reported by Pathways Commons. Our system may be reporting relations from other publications not considered by the Pathways Commons studies. Another hypothesis is that this low precision may be an effect of the different provenances for the data involved, which could also be affecting the Recall. The references come from actual GRN reported in Pathways Commons, whereas the data to train the model comes from a biomedical relation extraction dataset (BioRED) with relation pairs (e.g. gene–disease; chemical–chemical) at the document level, on a set of 600 PubMed abstracts, in which each relation has been labelled as describing either a novel finding or previously known background knowledge. BioRED has been assessed by benchmarking several existing state-of-the-art methods and results show that there is much room for improvement for the relation extraction task (F-score of 47.7%).
Regarding the high variability in the sizes of the knowledge bases, we want to highlight situations like this: when the reference size is larger than the size of the model, many of its events may be covered by events in the model with a relationship (such as association), that covers up a broad spectrum of possible interactions in the reference (like in-complex-with, controls-production-of, used-to-produce, among others) (see EVALUATION/Selenocysteine-synthesis). In such a situation, an event with a general relationship such as association will only be counted once, leaving other implicated events as false negatives, which negatively impacts the recall.
Nevertheless, achieving high recall under those conditions is particularly encouraging because it indicates that the system is effectively recovering, from a possibly considerably bigger collection of documents, information that coincides with well-curated, state of the art, reports of GRN. Pathways commons does not contain a manually adjusted set of positive examples, prepared to train an LLM, but human-oriented reports of conserved truths in biology. Therefore, we believe that these levels of recall represent a deeper confirmation of the effectiveness of the AI tools.
BioREx was trained with the summaries of papers, and that means that when PubTator predicts a relation between two biological objects, the LLM takes the whole abstract of the paper as the text that explains the relation. BioREx depends on PubMedBERT , a PreTrained Language Model (PLM) specialized on the PubMed abstracts. BERT based LLMs, are very good at learning the context that relates the entities in a sentence. We believe the BioREx’s false positive rate could be reduced by training the BioREx LLM at the sentence level, instead of the abstract level. The problem with such a change is the need for a training corpus properly tagged. We are taking steps in that direction regarding the NER and Normalization tasks. PubTator offers a very handy service as it offers in one place the normalization, ner and relation extractions tasks.
The PubTator's normalization of names does not always match the names of the objects provided as input to the system and that is normal; but this is more frequent in the case of small molecules than for genes and proteins as the metrics reveal. Table 6, Table 7, Table 8 and Table 9, show the metrics values for the experiments presented in Table 4 and 5, differentiated by whether or not they include small molecules, and they show a small difference when the reference does not include the last ones. We can say, however, that regardless of the case, the names of the objects of interest are generally not fully detected by PubTator and this, as expected, negatively impacts the quality of our models.
Evaluation of pairs-like relations generation in Biopatternsg with small molecules
Evaluation of pairs-like relations generation in Biopatternsg without small molecules
Evaluation of relations extraction in Biopatternsg with small molecules
Evaluation of relations extraction in Biopatternsg without small molecules
6. Conclusion
We have tested the feasibility of a methodology that combines generative AI and logical AI. We are leveraging the capacity of generative AI to address the task of entity and relations extraction. With respect to logical AI, we developed prolog scripts to infer biological object's identities from knowledge bases automatically organized, in addition to handling of constraints, which guide the inference of regulatory pathways and subnetworks.
The experiment also suggests the need to model the task of relation extraction at the sentence level, as opposed to the current modeling approach, developed at the abstract level; that granularity is important to improve the biological understanding about the knowledge domain on consideration. The automatic modeling of the knowledge bases with object's names and synonyms, and their identities, requires improvements. And this requires devising ways to map identifiers from a service like Uniprot to another like MeSH. So far, we only inferred regulatory pathways using the object's identities coming from MeSH. In the near future, we will perform experiments with regulatory pathways restricted by the knowledge in the ontology trees provided by gene ontology and the metadata provided by Uniprot and PDB; we have not yet built GRNs with restrictions shaped by those services.
The results shown in Table 4 and Table 5 establish that an initial exploratory phase conducted by the researcher could help; the idea is to review manually a better alignment of the objects of interest with the names assigned to them by the PubTator’s LLMs. Therefore, future experimental tasks regarding Table 4 and Table 5 involve running experiments with a better-aligned list of objects, and measuring from there the impact that this may have on the results that we get. We did not proceed in that manner in this work, as our objective was to see how far our pipeline could go in modeling knowledge bases, without human intervention.
Results such as those described in Table 5 and Table 6 are possible even with the metrics shown in Table 4 and Table 5, given the wide variety of interactions that generative AI provides and that logical AI can leverage to yield valuable findings. Stage 1 of the pipeline guides the gathering of abstracts from PubMed, then the stage 2 shapes from them a corpus of information closely related to a research problem; the COVID-19 experiment presented here demonstrates the type of functionalities that we have implemented to leverage that kind of corpora. The COVID-19 experiment also shows that our pipeline defines a knowledge management system useful to formulate hypothetical scenarios; in addition to facilitating the modeling of the specific domain of biology in which the researcher is working.