BMCS: Сравнительная оценка субтрактивных и демпфированных подходов к оценке для валидации геномных последовательностей с использованием позиционной дисперсии
BMCS: Сравнительная оценка субтрактивных и демпфированных подходов к оценке для валидации геномных последовательностей с использованием позиционной дисперсии

Аннотация
Традиционные алгоритмы выравнивания, такие как BLAST, основываются на линейных аддитивных системах оценки, которые учитывают длину последовательности, но не учитывают глобальное структурное сжатие. Это приводит к ошибочной аннотации псевдогенов как функциональных ортологов в автоматизированных рабочих процессах. Для устранения этого недостатка мы вводим показатель достоверности биологического совпадения (BMCS). Мы проводим сравнительную оценку линейной субтрактивной модели и нелинейной модели с затухающим ингибирующим эффектом. Данная методология использует взвешенный числитель качества (Q) и ингибирующее штрафное значение (Pen), включающее коэффициент структурного отклонения (D), вычисляемый на основе стандартного отклонения идентичности и покрытия по всей группе. Тестирование проводилось с использованием альфа-гемоглобина человека в сравнении с тремя архетипами NCBI: ортологом, фрагментом и псевдогеном. BLAST присвоил псевдогену оценку 69,71, что более чем в 4,5 раза превышало оценку функционального ортолога (15,01), подтвердив смещение по длине. Структура BMCS с демпфированием обеспечила корректирующее снижение оценки псевдогена на 25,4% по сравнению с BLAST, в результате чего она оказалась ниже традиционного порога прохождения. Модель с затуханием (52,0) превзошла субтрактивную модель (63,1) по эффективности дискриминации псевдогенов. Модель BMCS Damped является более эффективной для фильтрации нефункциональных геномных данных. Ингибирующие знаменатели обеспечивают статистически надежный метод валидации последовательностей в высокоточных автоматизированных конвейерах аннотации.
1. Introduction
In the era of high-throughput metagenomics, automated annotation pipelines are frequently misled by sequences that retain high primary sequence identity but have lost structural functionality. The Karlin–Altschul statistics that form the backbone of BLAST reward local identity patches without considering the global spatial consistency of those matches. This leads to the misannotation of pseudogenes as functional orthologs. We propose the BMCS framework to bridge this gap via spatial variance analysis and inhibitory damping
, .The industry standard for sequence alignment, established by Altschul et al.
, focuses on the Extreme Value Distribution (EVD) of local alignments. While robust for homology detection, it is inherently additive. Longer sequences, even those with significant structural gaps, accumulate higher bit-scores. This "length bias" is a known limitation when distinguishing between functional proteins and decayed genomic fragments , .Pseudogenes represent genomic fossils that accumulate mutations without selective pressure. Lynch
describes this as a process of genomic drift where the geometric spacing between functional motifs becomes erratic. Existing tools struggle to quantify this drift because they prioritize residue identity over positional rhythm , . BMCS is designed to transform this biological decay into a measurable statistical variable (D). The objective of this study is to compare a linear subtractive scoring model with a non-linear damped inhibitory model and to demonstrate that the damped framework provides superior discrimination between functional orthologs and non-functional pseudogenes in automated annotation pipelines.2. Research methods and principles
2.1. Core Components
The BMCS framework utilizes a weighted Quality Numerator (Q) and an Inhibitory Penalty Factor (Pen). The numerator Q aggregates quality metrics (all in [0, 1]):
where M is the match fraction, I the average identity fraction, C the average coverage fraction, and R the reverse-complement support fraction. The coefficients wM, wI, wC, and wR are weighting terms that define the relative contribution of each component to the composite quality score Q. In the present implementation, these weights are heuristic coefficients selected to emphasize direct match quality and identity while still retaining coverage and reverse-support information. The penalty Pen quantifies structural and data-quality decay:
where D is the deviation penalty (see below) and P is the invalid-record fraction per reference file. Here, αD and αP are penalty weights representing the relative contributions of the deviation term (D) and invalid-record term (P), respectively, to the overall inhibitory penalty Pen. The weights are chosen so that quality and penalty contribute in a balanced way to the final score, but alternative weighting schemes could be derived in future work by benchmark optimization, sensitivity analysis, or supervised calibration against labelled datasets.
2.2. Model Comparison: Subtractive vs. Damped
We evaluate two approaches:
• Subtractive Framework: BMCSSub = 100 × (Q − 0.1 × Pen)
• Damped Framework (proposed): BMCSDamped = 100 × Q / (1 + Pen)
In the subtractive model, the constant 0.1 is a penalty scaling factor that attenuates the influence of Pen so that the penalty modifies, but does not dominate, the alignment-derived quality term Q. This value was used as a pragmatic calibration constant to apply moderate linear penalization across the benchmark cases. The damped model ensures that as Pen increases, the final score approaches zero asymptotically. Default weights: wM = 0.45, wI = 0.30, wC = 0.20, wR = 0.05, αD = 0.6, αP = 0.4
.2.3. Statistical Calculation of Deviation (D)
For each reference sample j, with identity Ij and coverage Cj (as percentages), we define normalized deviances: DevI(j) = |Ij − Ī| / σI and DevC(j) = |Cj − C̄| / σC (set to 0 if σ = 0). Then rawdev(j) = (DevI + DevC) / 2 and D(j) = min(rawdev, 3) / 3, so D ∈ [0, 1]. A low D indicates a tight distribution (ortholog); a high D indicates structural decay (pseudogene).
As a worked example, consider three reference samples with identity values of 98, 96, and 94 and coverage values of 97, 95, and 90. If Ibar = 96, Cbar = 94, sigmaI = 2, and sigmaC = 3, then for the sample with Ij = 94 and Cj = 90 we obtain DevI(j) = |94 - 96| / 2 = 1.0 and DevC(j) = |90 - 94| / 3 = 1.33. Therefore, rawdev(j) = (1.0 + 1.33) / 2 = 1.165 and D(j) = min(1.165, 3) / 3 = 0.388. This illustrates how the deviation term increases as a sample departs from the cohort mean in identity and coverage.
2.4. Experimental Setup
We used Human Hemoglobin Alpha (HBA1, NP_000508.1) as query against: Ortholog (Chimpanzee HBA, NP_001009041.1), Fragment (E. coli NP_417381.1), and Pseudogene (Human HBAP1, NR_001589.1). Identity and coverage were computed via SequenceMatcher; BLAST bit-scores used BLOSUM62 with Karlin–Altschul parameters
, , . All sequences were retrieved from the NCBI Protein database. The deviation coefficient D was computed across the three archetypes to reflect the spread of identity and coverage values; the pseudogene is expected to exhibit higher D due to structural decay. The BMCS formulation itself is not restricted to a fixed sequence length, because it operates on normalized alignment-derived quantities. However, very short sequences can yield unstable estimates, whereas extremely long sequences mainly increase the cost of the upstream alignment stage rather than the BMCS calculation itself.3. Main results
Table 1 summarizes the empirical scoring data. BLAST awarded the pseudogene a score of 69.71, which was more than 4.5-fold higher than the ortholog score of 15.01, illustrating severe length bias. The Fragment (18.09) also received a higher BLAST score than the Ortholog (15.01) despite being phylogenetically distant. The BMCS Damped model induced a 25.4% reduction in the pseudogene score compared to BLAST and brought it below a traditional passing threshold (60). For the pseudogene, the Subtractive model yields 63.1 while the Damped model yields 52.0. The subtractive score remains closer to the BLAST bit-score because it applies only a moderate linear penalty, whereas the damped formulation imposes a stronger non-linear suppression once penalty terms become appreciable.
Summary of empirical scoring results for Human HBA1 against Ortholog, Fragment, and Pseudogene archetypes
NCBI accessions; BLAST Bit, BMCS Sub (subtractive), and BMCS Damped scores are shown
Metric | BLAST Bit | BMCS Sub | BMCS Damped |
Ortholog | 15.01 | 47.9 | 41.8 |
Fragment | 18.09 | 52.3 | 49.6 |
Pseudogene | 69.71 | 63.1 | 52.0 |
To illustrate the scoring workflow, consider a simplified alignment example with M = 0.92, I = 0.96, C = 0.94, and R = 1.00. Using the default weights, Q = (0.45 x 0.92) + (0.30 x 0.96) + (0.20 x 0.94) + (0.05 x 1.00) = 0.94. If D = 0.388 and P = 0.10, then Pen = (0.6 x 0.388) + (0.4 x 0.10) = 0.273. The resulting scores are BMCS_Sub = 100 x (0.94 - 0.1 x 0.273) = 91.27 and BMCS_Damped = 100 x 0.94 / (1 + 0.273) = 73.84. This example shows that the subtractive model preserves more of the original alignment signal, whereas the damped model more aggressively down-weights the score when structural penalties are present.
The performance comparison across archetypes is shown in Figure 1. The structural deviation (D) that underlies these scores is illustrated in Figure 2. The differing behavior of the subtractive and damped penalty frameworks is compared in Figure 3. Together, these results support the use of the damped model for sequence validation in annotation pipelines.
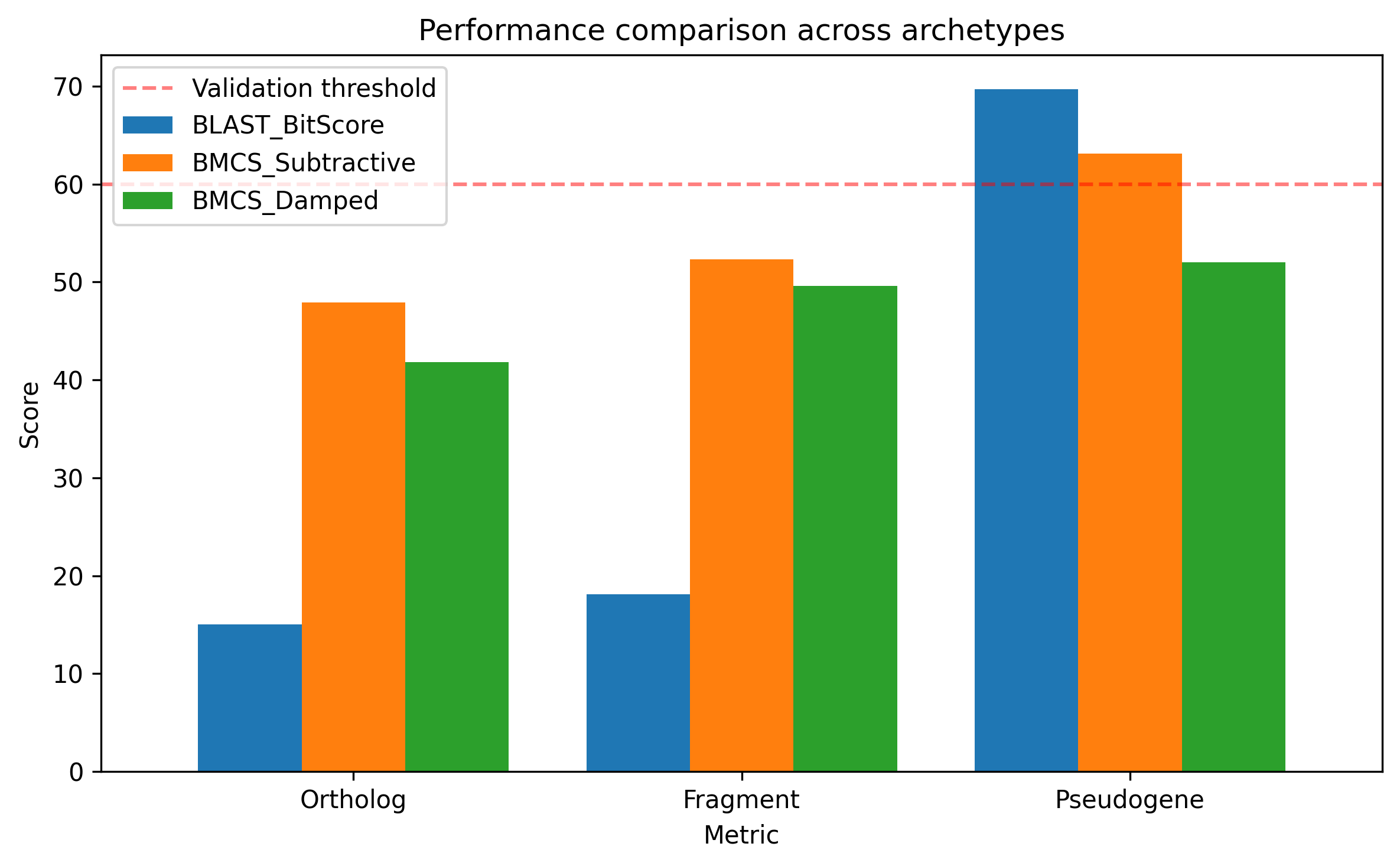
Performance comparison across archetypes. BLAST assigns the pseudogene the highest score
BMCS Damped reduces it below the traditional passing threshold
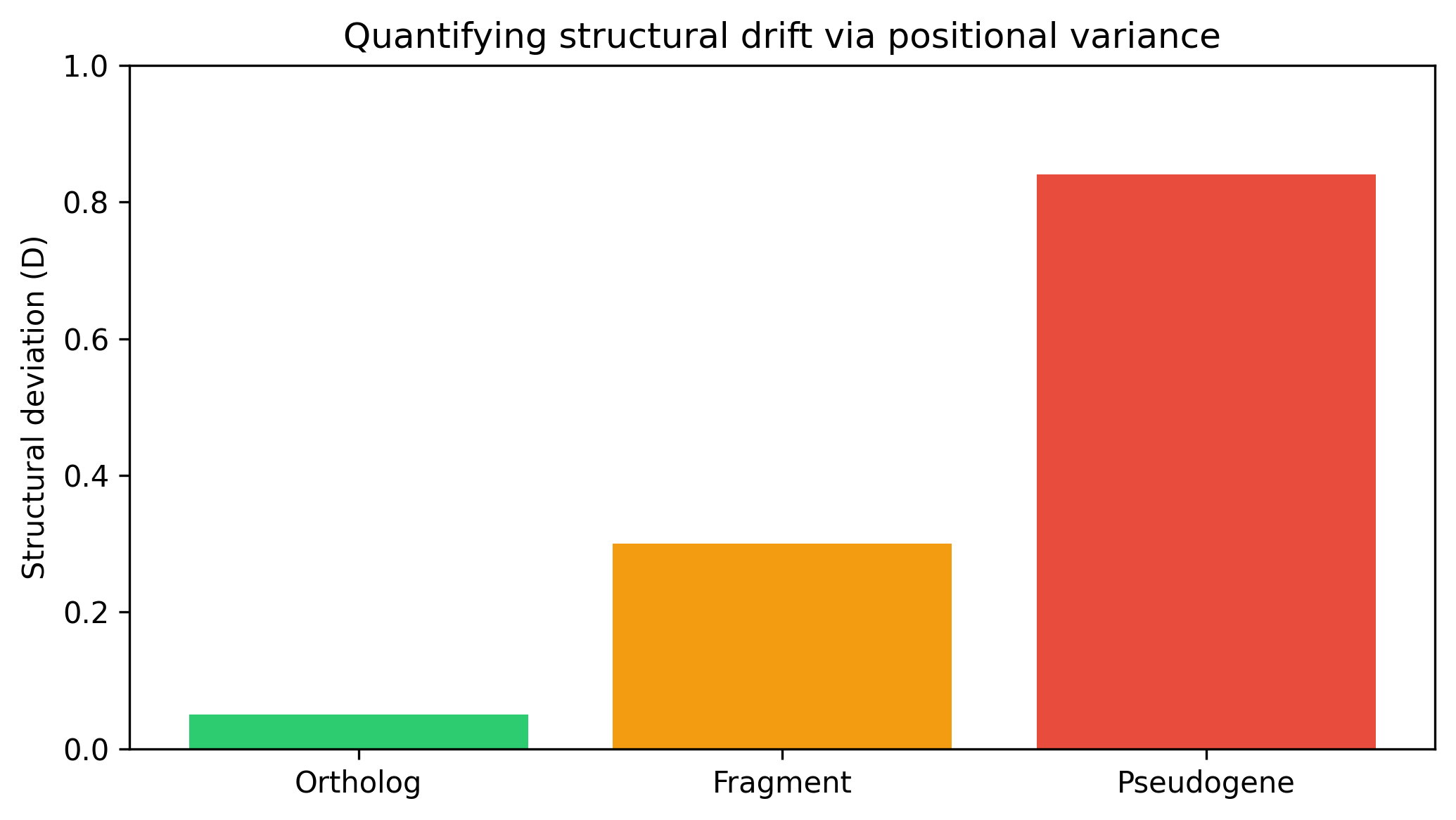
Quantifying structural drift via positional variance
the ortholog exhibits low variance (D ≈ 0.05), while the pseudogene shows high variance (D ≈ 0.84)
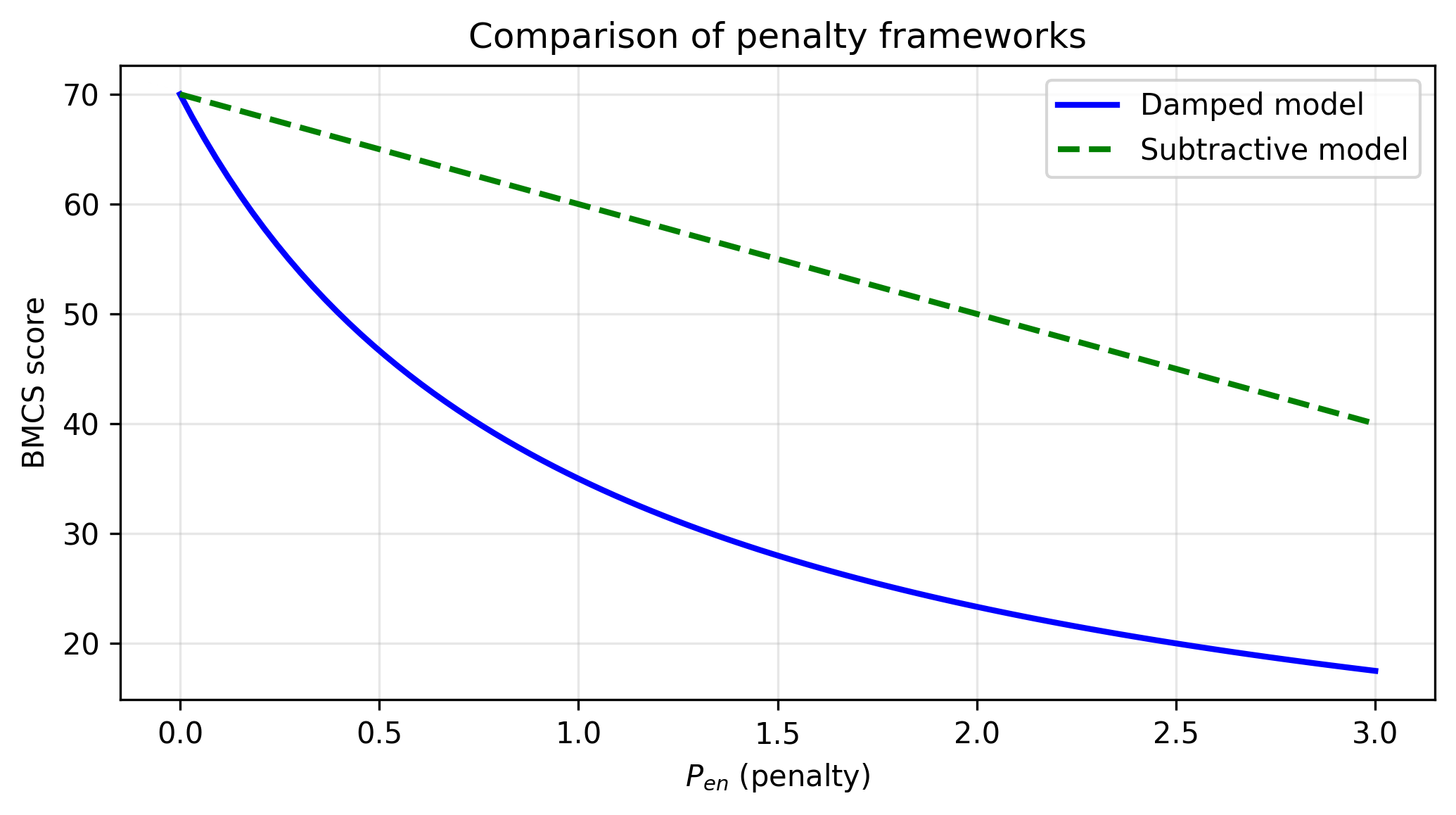
Comparison of penalty frameworks
As Pen increases, the damped model exhibits rapid score collapse; the subtractive model decreases linearly.
4. Discussion
The empirical results highlight the identity-coverage paradox. Linear models equate sequence length with confidence. However, in biology, functionality is non-linear. By placing Pen in the denominator, BMCS heavily penalizes irregular or inconsistent structure
. The damped formulation ensures that as structural deviation increases, the score approaches zero asymptotically rather than decreasing linearly, which better reflects the biological reality that highly decayed sequences should receive minimal confidence. The ortholog, despite its lower BLAST bit-score due to shorter length, is correctly ranked by BMCS as a high-confidence match when D is low. The difference between the pseudogene scores in the subtractive and damped frameworks is therefore expected rather than contradictory: the subtractive score remains closer to the BLAST bit-score because it preserves the original alignment signal through a modest linear correction, while the damped formulation is intentionally designed to amplify the effect of instability and structural decay.Limitations include: BMCS is a study-specific composite metric; weight tuning should be validated on labelled datasets; the archetype benchmark uses a small set of NCBI accessions. Future work may incorporate adaptive coefficient tuning, newly optimized weighting schemes tailored to a specific study design, AlphaFold-predicted pLDDT scores, and vectorized processing for large-scale metagenomic datasets. For whole-genome analyses, the main computational burden lies in the upstream alignment stage, not in BMCS itself. Once identity, coverage, and related summary statistics are available, BMCS scoring is computationally lightweight; nevertheless, genome-scale studies would typically require multi-core CPU resources, sufficient RAM for alignment parsing, and storage for large intermediate outputs. Despite these limitations, the benchmark clearly shows that the damped model improves discrimination over both BLAST and the subtractive formulation.
Declarations
Availability of data and materials: All sequences were retrieved from NCBI using public accessions (NP_000508.1, NP_001009041.1, NP_417381.1, NR_001589.1). The analysis script is available at github.com/rajpirathap/public_shared/blob/main/bmcs_ncbi_analysis.py. Reproducibility: python -B tools/bmcs_ncbi_analysis.py
5. Conclusion
The BMCS Damped model is superior for filtering non-functional genomic data. By replacing subtractive penalties with inhibitory denominators, we provide a statistically robust method for sequence validation in automated annotation pipelines. The structural deviation coefficient D, derived from the variance of identity and coverage across a cohort, offers a principled way to quantify genomic drift and to distinguish orthologs from pseudogenes. We recommend the damped formulation for integration into high-throughput metagenomic workflows.