ПРИМЕР ЦЕЛЕВОГО ПОДХОДА К РАСПОЗНАВАНИЮ РОДА КОРОНАВИРУСОВ С ПОМОЩЬЮ МЕТОДА ГЛАВНЫХ КОМПОНЕНТ
ПРИМЕР ЦЕЛЕВОГО ПОДХОДА К РАСПОЗНАВАНИЮ РОДА КОРОНАВИРУСОВ С ПОМОЩЬЮ МЕТОДА ГЛАВНЫХ КОМПОНЕНТ

Аннотация
На примере распознавания рода коронавирусов представлен оригинальный подход к определению таксономии вирусов в метагеномных исследованиях. Подход относится к целевым (таргетным) методам по распознаванию вирусов на основе частот кодонов в N-гене белка нуклеокапсида. Используемый подход основан на нестандартном применении метода главных компонент к обучающим выборкам векторов частот кодонов для N-генов коронавирусов разных родов. Показана репрезентативность предложенной обучающей выборки. Продемонстрирована возможность значительного уменьшения параметров распознавания путем сокращения размерности пространства векторов частот кодонов N-гена. Предлагаемый подход к определению таксономической принадлежности вирусов относится к группе методов без выравнивания, развиваемых в последние десятилетия для выявления родства и эволюции видов вирусов по их секвенированным геномам.
1. Введение
Таксономия вирусов, установленная и развиваемая Международным комитетом по таксономии вирусов ICTV , не только описывает и систематизирует наши знания об известных вирусах, но также способствует разработке вакцин и методов лечения вирусных инфекций. В случае обнаружения нового патогенного вируса определение его вида, рода и семейства ускоряет выбор возможной терапии и вакцинации, особенно если средства и способы борьбы против родственных ему вирусов уже существуют.
Благодаря современным технологиям классифицировать вирусы и определять эволюционные связи между ними возможно в результате сравнительного анализа их геномов. Быстрое развитие технологий секвенирования и рост метагеномных данных создают запрос на создание новых вычислительных методов, программ для анализа и классификации вирусов. С переходом к революционной нанопоровой технологии (Oxford Nanopore) секвенирования третьего поколения с длинным прочтением (LRS — long-read sequencing) открываются новые возможности вирусной метагеномики , . В отличие от высоко производительной технологии NGS (Illumina), известной как секвенирование с коротким прочтением (SRS — short-read sequencing), LRS технология позволяет значительно быстрее и надежнее определять области генома, содержащие тандемные повторы, и реконструировать полные геномы за одно прочтение . Технология LRS способствует классификации вирусов с высоким разрешением и точному определению важных генетических элементов. Кроме того, её существенным преимуществом является возможность идентифицировать химические модификации нуклеотидов, возникающие на уровне эпигенетических процессов.
Основные подходы к классификации метагеномных данных (идентификации вирусов) можно условно разделить на четыре основные группы : выравнивание; статистические и скрытые марковские модели (HMM); идентификация вирусов на основе машинного обучения на основе частот k-меров (небольших последовательностей из k нуклеотидных или аминокислотных остатков); гибридные методологии, которые сочетают в себе несколько стратегий.
Одними из первых, и широко используемых в настоящее время в метагеномике, подходов к таксономии являются методы определения сходства путем выравнивания с референсными геномами из баз данных, например, RefSeq . Программа BLAST является самым известным инструментом в этой группе. Специализированный портал NCBI Virus предоставляет BLAST-интерфейс для анализа вирусных последовательностей, сравниваемых с тщательно отобранными метаданными о вирусах, в том числе и с коронавирусами рода Betacoronavirus. В настоящее время широко используются программы множественного выравнивания такие, как MUSCLE , MAFFT и др. По результатам работы этих программ методами филогении (наиболее часто методом правдоподобия ) строятся дендрограммы вирусных таксонов. В случае, когда денрограммы строятся на основе выравненных последовательностей, количественные различия определяют меру расстояния между ними.
Выравнивание последовательностей и анализ k-меров, требуют очень больших вычислительных затрат, что осложняет их применение для оценки огромных массивов данных. Потому в последние два десятилетия разрабатываются различные методы без выравнивания, обладающие достаточной эффективностью , , . Методы без выравнивания для сравнения и классификации последовательностей извлекают и используют их характерные признаки или шаблоны.
Наши работы по распознаванию рода коронавирусов (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus) также направлены на создание новых подходов к распознаванию таксономической принадлежности вирусов без сравнительного выравнивания их геномных последовательностей относительно известных геномов вирусов. С этой целью в настоящей работе анализируемый вирус характеризуется вектором частот кодонов одного из его генов, кодирующего вирусный белок нуклеокапсида.
Ранее для распознавания рода коронавирусов на основе частот кодонов в генах нами рассматривался вариантный подход, который использовал как отдельные структурные (S-, M-, N- гены) и неструктурные гены (объединенные в единой ORF1ab), так и их различные комбинации-варианты . Всего было рассмотрено шесть вариантов. Отметим, что ORF1ab содержит, в частности, ген РНК-зависимой РНК-полимеразы (RdRp), обычно используемый для разделения видов, родов и семейств коронавирусов .
Анализируемому геному коронавируса при вариантном подходе соответствовала вариантная строка, компоненты которой содержали результат распознавания рода коронавируса по каждому варианту. Мозаичность компонент вариантных строк отражала рекомбинантные процессы в геномах. Наиболее эффективное распознавание рода продемонстрировал вариант, использующий только ORF1ab. Распознавание рода коронавируса при рассмотрении частот кодонов структурных генов оказалось чуть менее эффективно, с уровнем достоверности (чувствительности) не менее 95%.
Вариантный подход использовал сравнение частот кодонов в генах анализируемого вирусного генома с усредненными частотами кодонов в соответствующих генах прототипных штаммов каждого рода коронавирусов, то есть вирусов, рассматриваемых Международным комитетом по таксономии вирусов ICTV в качестве типичных представителей вирусного таксона. При этом среди структурных генов распознавание на основе N-гена показало недостаточно высокую чувствительность только для одного рода Alphacoronavirus.
Хотя N-ген имеет достаточно малую длину — 1200 нукл. (для сравнения S-ген имеет длину порядка 3700 нукл.), ранее он успешно использовался для построения филогенетических дендрограмм при изучении географии распространения коронавирусов и в молекулярно-эпидемиологическом анализе вариантов вируса SARS-CoV-2 . В работе по распознаванию рода и подрода коронавируса также использовался N-ген белка нуклеопротеина, частоты кодонов которого из анализируемого генома сравнивались с частотами кодонов в N-генах индивидуальных прототипных штаммов подродов коронавирусов. Такой подход был назван типологическим , .
В работе было показано, что при типологическом подходе распознавание на основе N-гена наиболее эффективно (в сравнении с S-геном и ORF1ab). Поэтому N-ген белка нуклеокапсида был выбран в качестве целевого (таргетного) гена в дальнейшем анализе.
Сложность типологического подхода обусловлена необходимостью сравнения с большим числом прототипных штаммов. В этом его сходство с методами выравнивания, которые также требуют сравнения с множеством эталонов.
Для упрощения задачи распознавания рода коронавирусов нами было выполнено сравнение вектора частот кодонов в анализируемом N-гене с усредненными по родам векторами частот кодонов в N-генах из обучающей выборки. Однако такой подход показал слишком низкую эффективность распознавания рода коронавируса. Поэтому в работе для распознавания рода коронавирусов был предложен другой подход, в котором особым образом использовался метод главных компонент для векторов частот кодонов в анализируемых N-генах. В этом подходе на основе обучающей выборки для каждого рода коронавирусов создаются процедуры преобразования векторов частот кодонов в N-генах. Для фиксированного рода процедура преобразования выполняется следующим образом. Сначала на основе векторов частот кодонов в N-генах из обучающей выборки рассматриваемого рода коронавирусов вычисляется усредненный вектор частот кодонов в этом роде. На его основе осуществляется трансформация каждого вектора частот кодонов рода, и для каждого рода коронавирусов создается соответствующая выборка трансформированных векторов частот кодонов. Далее к выборке трансформированных векторов частот кодонов N-генов каждого рода применяется метод главных компонент, для которых найдены их дисперсии. Таким образом, на основе обучающей выборки для анализируемого вектора частот кодонов N-гена созданы фиксированные процедуры его преобразования, зависящие от рода коронавирусов.
Согласно процедуре предполагаемого рода, анализируемый вектор частот кодонов N-гена коронавируса сначала трансформируется и, затем, для трансформированного вектора вычисляются главные компоненты. В результате для предполагаемого рода коронавирусов вычисляется сумма квадратов значений главных компонент, нормированных на соответствующие им дисперсии. Таким образом, для анализируемого коронавируса вычисляются такие суммы для возможных распознаваемых родов. На основе минимальной суммы выбирается род анализируемого коронавируса.
Описываемый выше способ распознавания рода коронавирусов основан на предположении о том, что для каждого рода векторы частот кодонов в N-генах коронавирусов могут быть достаточно близки к различным нормальным многомерным распределениям типичным для каждого рода коронавирусов. Эксперименты показали, что, начиная с некоторой размерности пространства векторов частот кодонов N-гена, достигался уровень чувствительности 95% (и выше) в распознавании рода коронавируса для обучающей и тестируемых выборок. Однако для тестируемой выборки в работе наблюдались следующие явления. Тестируемая выборка показывала приемлемый уровень чувствительности (95%) распознавания рода Betacoronavirus только для достаточно высокой размерности (n=28) пространства векторов частот кодонов в N-генах. Кроме того, для рода Deltacoronavirus, начиная с размерности 16 происходит существенное снижение уровня чувствительности к распознаванию этого рода. По-видимому, такие явления обусловлены недостаточной репрезентативностью обучающей выборки.
В настоящей работе увеличен размер обучающей выборки. Показано, что приемлемый уровень эффективности распознавания рода коронавирусов достигается, начиная с достаточно низкой размерности пространства анализируемых векторов частот кодонов в N-генах и стабильно возрастает с ростом этой размерности.
Таким образом, в настоящей работе на примере распознавания рода коронавируса по N-гену белка нуклеокапсида демонстрируется возможность успешного применения метода главных компонент для классификации вирусов без выравнивания их полных геномов, с опорой только на выделенный целевой ген. Предлагаемый подход может существенно упростить распознавание последовательностей геномов, и избежать ошибок идентификации, обусловленных частыми рекомбинациями между вирусными геномами. Кроме того, дополнительно упростить анализ возможно путем значительного уменьшения размерности векторов частот кодонов, характеризующих целевой ген.
2. Методы и принципы исследования
В предыдущей работе , посвященной распознаванию рода коронавирусов, основанному на методе главных компонент, объем обучающей выборки N-генов был существенно меньше объема тестируемой. В настоящей работе объем обучающей выборки значительно увеличен, особенно для родов Deltacoronavirus и Gammacoronavirus. Численный состав по родам для обучающей и тестируемой выборок N-генов коронавирусов в настоящей работе, соответственно, составлял: для Alphacoronavirus (α-CoV) — 1281 и 251, для Betacoronavirus (β-CoV) — 2126 и 1647, для Deltacoronavirus (δ-CoV) — 244 и 80, и для Gammacoronavirus (γ-CoV) — 406 и 135. Обучающая и тестируемая выборки различались между собой в среднем на 91%.
Исходные последовательности N-генов были получены из базы данных GenBank . Порядок компонент в векторах частот кодонов соответствовал нумерации кодонов, упорядоченных по убыванию частот встречаемости в N-генах прототипных штаммов для родов коронавирусов, GenBank коды доступа которых приведены в работе . Таблица 1 представляет список упорядоченных таким образом кодонов, за которыми закреплены соответствующие номера. Последние пять кодонов, включая кодоны терминации, с самыми низкими частотами были исключены из анализа. Убывание частоты встречаемости кодона происходит в соответствии с ростом его порядкового номера в таблице 1.
Нумерованный список кодонов аминокислот в соответствии с убыванием их частоты встречаемости в N-генах прототипных штаммов подродов коронавирусов
по ист. [23]
1 aag | 2 gat | 3 aaa | 4 gct | 5 ggt | 6 aat | 7 cct | 8 caa | 9 cca | 10 tct |
11 cag | 12 act | 13 gga | 14 gaa | 15 aga | 16 gca | 17 gtt | 18 gac | 19 ttt | 20 cgt |
21 tca | 22 aac | 23 att | 24 ctt | 25 gag | 26 aca | 27 ggc | 28 tgg | 29 agt | 30 gcc |
31 ttc | 32 atg | 33 tat | 34 ccc | 35 cgc | 36 acc | 37 tac | 38 gtg | 39 ttg | 40 gtc |
41 cat | 42 agc | 43 agg | 44 tcc | 45 gta | 46 ctg | 47 ggg | 48 atc | 49 ctc | 50 cta |
51 gcg | 52 ccg | 53 ata | 54 cga | 55 cac | 56 tta | 57 tcg | 58 acg | 59 cgg |
|
Кратко опишем метод распознавания рода коронавирусов на основании метода главных компонент.
Каждый N-ген характеризуется вектором частот кодонов в виде
Для анализируемого вектора частот коронавирусов опишем его трансформацию, соответствующую каждому роду коронавирусов, где
Для каждого рода по всем генам его обучающей выборки рассчитываются средние частоты встречаемости каждого рассматриваемого кодона. Тем самым для рода X создается вектор средних частот
На диагонали матрицы DX стоят собственные значения матрицы CX, являющиеся дисперсиями соответствующих главных компонент. Кроме того, QXT — транспонированная матрица (для QX), являющаяся матрицей преобразований к главным компонентам трансформированных многомерных векторов рода X.
Опишем процедуру распознавания рода анализируемого вектора частот
3. Результаты и обсуждение
Эффективность распознавания рода коронавирусов в предлагаемом подходе оценивалась с помощью двух параметров: чувствительности и специфичности распознавания. Согласно определению , чувствительность — отношение количества истинно положительных результатов к общему количеству объектов с признаком, умноженное на 100%; специфичность — отношение количества истинно отрицательных результатов к общему количеству объектов без признака, умноженное на 100%.
3.1. Распознавания рода коронавирусов в зависимости от объема обучающей выборки
Рассмотрим особенности в распознавании рода коронавирусов, проявившиеся на тестируемой выборке N-генов в работе , которые показывает рисунок 1. Как отмечалось во Введении, приемлемый уровень чувствительности (95%) в распознавании рода Betacoronavirus достигается, начиная с достаточно высокой размерности (n=28) пространства векторов частот кодонов N-гена. Одновременно с улучшением распознавания рода Betacoronavirus, для рода Deltacoronavirus, начиная с размерности n=21, происходит существенное снижение уровня чувствительности в распознавании этого рода. Такие явления говорят о недостаточной репрезентативности обучающей выборки в работе . Поэтому в настоящей работе объём обучающей выборки N-генов был значительно увеличен для всех родов коронавирусов.
![Чувствительность распознавания родов коронавирусов (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus) для N-генов (представленных векторами частот кодонов), соответствующая обучающей выборке в работе [21]](/media/images/2026-06-25/ded9ca59-3e24-45d5-be26-ed53dd598648.png)
Чувствительность распознавания родов коронавирусов (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus) для N-генов (представленных векторами частот кодонов), соответствующая обучающей выборке в работе [21]
доля N-генов с правильно распознанным родом показана в зависимости от размерности n рассматриваемого пространства векторов частот кодонов; нумерация компонент вектора сопровождается указанием кодона, частота которого соответствует компоненте; порядок следования кодонов соответствует их списку в таблице 1
Таким образом, увеличение объема обучающей выборки привело к отсутствию негативных особенностей в чувствительности предлагаемого подхода и его высокой специфичности при распознавании.
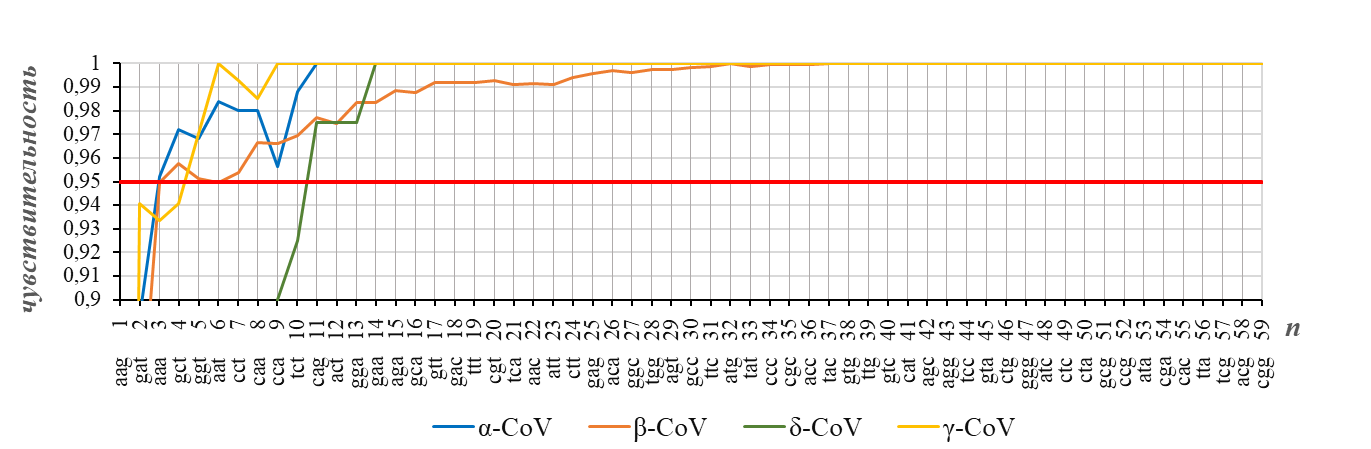
Чувствительность распознавания родов коронавирусов (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus) для N-генов (представленных векторами частот кодонов), достигаемая с помощью обучающей выборки, используемой в настоящей работе
доля N-генов с правильно распознанным родом показана в зависимости от размерности n рассматриваемого пространства векторов частот кодонов; рядом с номером компоненты вектора указан кодон, частота которого определяет эту компоненту; порядок следования кодонов соответствует их списку в таблице 1
Если обучающая выборка может считаться репрезентативной, то мы вправе ожидать, что результат распознавания с ее помощью любой тестируемой выборки не должен кардинально отличаться от результата, который достигается, когда обучающая выборка распознает саму себя. Рисунок 3 показывает, как обучающая выборка, используемая в настоящей работе, распознает себя в качестве тестируемой.
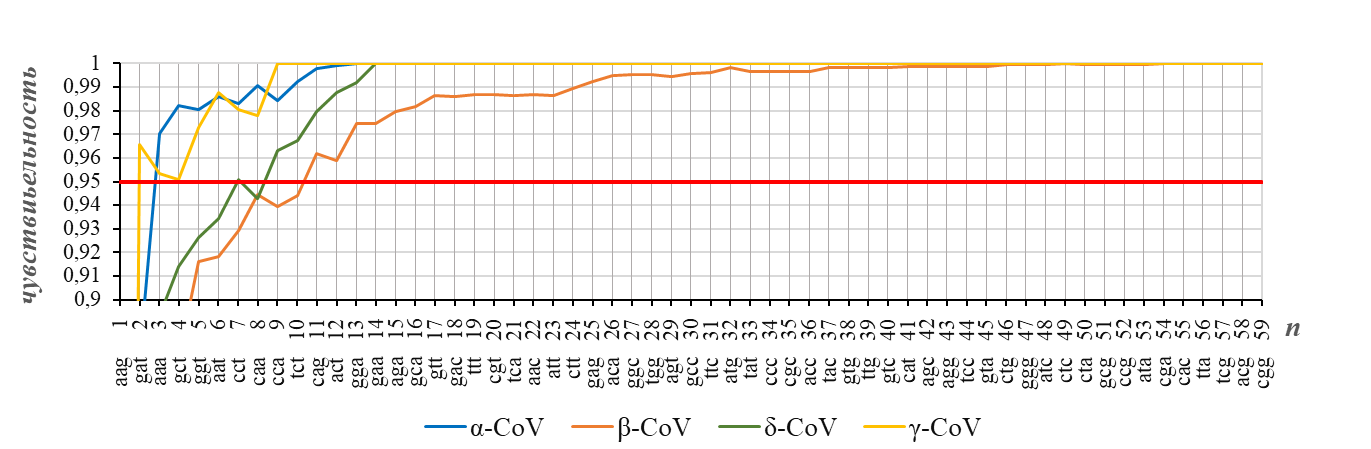
Чувствительность распознавания родов коронавирусов (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus) для N-генов (представленных векторами частот кодонов), полученные для обучающей выборки, используемой в настоящей работе, в случае, когда она одновременно является и тестируемой
доля N-генов с правильно распознанным родом показана в зависимости от размерности n рассматриваемого пространства векторов частот кодонов; рядом с номером компоненты вектора указан кодон, частота которого определяет эту компоненту; порядок следования кодонов соответствует их списку в таблице 1
Таким образом, из рисунка 2 и рисунка 3 следует, что для эффективного распознавания рода коронавирусов можно ограничиться достаточно низкой размерностью (n=12) пространства векторов частот кодонов в N-гене.
4. Заключение
В настоящей работе представлен пример использования оригинального подхода к распознаванию геномов коронавирусов, относящийся к методам целевого (таргетного) распознавания без выравнивания. Подход исходит из оценки частот встречаемости кодонов в таргетном гене (в настоящей работе это N-ген белка нуклеокапсида). Подход основан на использовании метода главных компонент и требует репрезентативной обучающей выборки, наличие которой показано в настоящей работе. Кроме того, отмечена возможность значительного уменьшения параметров распознавания путем сокращения размерности пространства векторов частот кодонов N-гена.