Анализ влияния мутаций на аффинность связывания в комплексах ACE2 и RBD S-белка коронавирусов
Анализ влияния мутаций на аффинность связывания в комплексах ACE2 и RBD S-белка коронавирусов
Аннотация
Методы, разрабатываемые для определения аффинности, имеют как и прикладное значение при поиске и разработке новых лекарств, так и значение для фундаментальной биофизики – поиск основных паттернов взаимодействия и автоматизированное описание систем интерфейсов белковых структур. В данной работе произведена оценка экспериментально полученных структур 30 комплексов рецептора ACE2 (Ангиотензинпревращающий фермент 2) с RDB (S-белок мутантного и дикого типа коронавирусов SARS-CoV и SARS-CoV-2, включая многочисленные мутантные формы последнего). Также проведен детальный структурный анализ, выявлены статистически значимые основные структурные элементы, вносящие высокий вклад в аффинность для комплексов с сильным связыванием (Kd < 30 nM). Полученная модель позволяет по-новому взглянуть на присутствие и вклад отдельных атомных групп в аффинность.
1. Введение
Проникновение вируса SARS-CoV-2 в организм человека начинается со взаимодействия RBD-домена его S-белка с белком ACE2, выполняющим роль рецептора. С начала пандемии были описаны сотни тысяч вариантов вируса, которые в настоящее время объединены, согласно сервису Nextstrain.org , в 26 клад. Каждый вариант вируса отличается от референсного варианта наличием уникального набора мутаций. Сервис Nextstrain.org рассчитывает и предоставляет данные о скорости накопления таких мутаций, причем скорость накопления мутаций планомерно увеличивается. Так, в период с декабря 2019 по ноябрь 2021 года эта скорость составляла 23 замены в год, а затем выросла до 50 замен в год, что совпало по времени с появлением высокопатогенного штамма Омикрон. Основным накопителем мутаций, в силу своего размера и функциональной важности является S-белок. Помимо этого, свободные S1-субъединицы белка шипов вируса SARS-CoV-2 могут действовать в качестве фактора патогенеза COVID-19 . Таким образом, наблюдения за накоплением мутаций подтверждают, что мутации в S-белке циркулирующих вариантов SARS-CoV-2 накапливаются со значительной скоростью, которая, вероятно, будет увеличиваться. Причиной этого является избирательное давление со стороны иммунитета хозяина, приобретенного в результате предыдущих инфекций и/или прививок, которое продолжает стимулировать быструю эволюцию .
В настоящее время в банке данных белковых структур PDB содержится более сотни структур комплексов hACE2-RBD SARS CoV-2, причем как нативного ACE2 и исходной формы RBD, так и их многочисленных мутантных форм. Список включает в себя штаммы альфа, бета, гамма, дельта, каппа, омикрон, а также многие другие варианты. Кроме того, имеются данные о структурах комплексов hACE2-RBD SARS CoV. Столь обширные структурные данные позволяют провести анализ молекулярных взаимодействий между RBD и hACE2 и выявить типичные и уникальные контакты между остатками.
Из литературных данных следует, что наиболее значимыми для связывания являются три области (hot spot). Первая локализована вокруг LYS353, располагающегося на интерфейсе ACE2, и стабилизируется взаимодействиями, образованными остатками THR500, ASN501, GLN 498, TYR505 со стороны RBD и LYS353, ASP38, TYR41, GLN42, LEU45 и ASN330 ACE2. Мутации в вышеописанных позициях значимо влияют на аффинность связывания, так, например, выявлено, что замена аспарагина на тирозин в 501 позиции RBD у альфа штамма SARS-CoV-2 значительно увеличивает аффинность , . Вторая область ассоциирована с LYS31, стабилизируется контактами, образованными LEU455, GLU484, LYS417 RBD, которые взаимодействуют с ASP30 и непосредственно с LYS31 . Также в мутантах дикого штамма обнаружен GLN493, который усиливает взаимодействие в данной области. Третья область расположена вблизи N конца ACE2, с которым взаимодействует короткая петля RBM вдали от основного интерфейса, что обеспечивает более обширную область взаимодействий . Критически важными для стабилизации этой области являются следующие остатки RBD: ALA475, GLY476 и PHE486, первые два взаимодействуют с SER19, а третий с MET82 и LEU79 . При этом ранее показано, что мутации в позиции GLY476, а также ALA475 негативно сказываются на аффинности связывания .
Межмолекулярные взаимодействия в комплексе RBD-ACE2 подробно освещаются практически в каждой работе, посвящённой той или иной расшифрованной структуре (ссылки на 6LZG, 6M0J, 7EKH, 7LO4, 7WHH), однако в них практически не уделяется внимание молекулам воды, расположенным на интерфейсе взаимодействия или в его окрестности. Между тем, структуры комплексов RBD-ACE2, полученные методом рентгеновского структурного анализа с высоким разрешением (как правило, 2,5 Ангстрем или лучше), содержат большое число молекул кристаллизационной воды. Так, в структурах 6LZG, 6M0J, 7EKH, 7LO4 и 7WHH содержится 322, 80, 250, 132 и 131 молекул воды, соответственно. Большая часть этих молекул расположена в карманах на поверхности hACE2, однако заметное число находится и в окрестности интерфейса взаимодействия этого белка с RBD (рис. 1). Интересно отметить, что множества молекул воды, находящиеся в этих структурах, с одной стороны, пересекаются, что позволяет выявить наиболее консервативные сайты связывания воды, а с другой стороны, дополняют друг друга, практически не создавая стерических затруднений. Это наблюдение, вероятно, позволяет создать молекулярную модель интерфейса, содержащую в себе все возможные молекулы воды.

Рисунок 1 - Молекулы воды на интерфейсе взаимодействия RBD-ACE2 на примере структур 6LZG, 7EKH и 7LO4
Примечание: совмещение выполнено по остаткам, образующим интерфейс или прилегающим к ним. Среднеквадратичное отклонение структур по Са-атомам < 1 Ангстрема
При этом анализ интерфейса белок-белковых взаимодействий позволяет выявить ключевые пары остатков, которые вносят наиболее существенный вклад в связывание белков. Целью данной работы стал статистический анализ межатомных расстояний и выявление пар атомов, положительно и отрицательно влияющих на связывание.
2. Методы и принципы исследования
В результате анализа литературы были отобраны 30 структур комплекса ACE2 и RBD вирусов SARS CoV (3 структуры) и SARS CoV-2 (27 структур), включая исходный вариант, а также различные штаммы и инженерные варианты (приложение 1). Для комплексов авторами было измерено значение константы диссоциации (Кд). С целью последующего изучения комплексов они были разделены на два класса: низкоаффинные (Кд > 30 нМ, 11 комплексов) и высокоаффинные (Кд < 30 нМ, 19 комплексов).
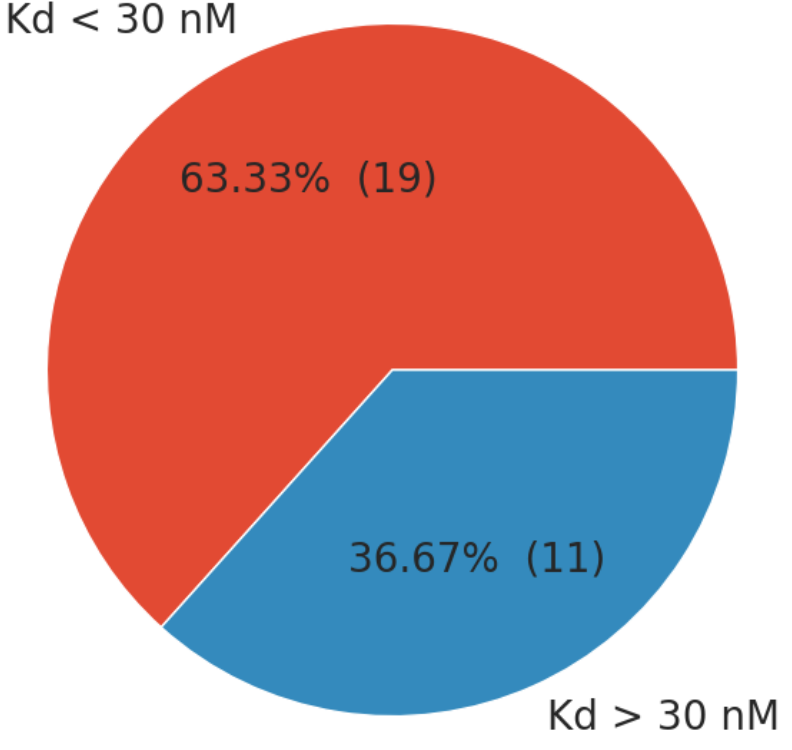
Рисунок 2 - Структура набора данных, всего 30 комплексов ACE2-RBD, 19 из которых были определены в группу высокой аффинности, 11 в группу низкой
Каждый из комплексов представлен в виде матрицы потенциальных контактов атомов групп, согласно типизации тяжелых атомов в силовом поле CHARMM (приложение 2), таких групп 15. Расчет расстояний производился с помощью программного пакета MDAnalysis. Вычислительный алгоритм учитывает возможность взаимодействия атомов в том случае, если атомы разных цепей находятся на заданных интервалах расстояний; эмпирически было установлено 4 таких интервала: 2-4, 4-5, 5-6, 6-10 Å.
Создание матриц расстояний: для каждого комплекса изначально таблица взаимодействий представляла вид, где по столбцам 15 групп атомов для цепи, соответствующей ACE2, по строкам – 15 групп атомов для цепи RBD соответственно, в значениях ячеек отражено количество связей на заданном интервале расстояний (пусть идентификатор PDB структуры 6LZG, а диапазон расстояний 6-10 Å) (табл. 1).
Таблица 1 - Таблица взаимодействий для структуры 6LZG. По вертикали атомы RBD, по горизонтали - ACE2, диапазон расстояний 6-10Å
CHARMM atom-types | NH1 | O | … | NH3 |
NH1 | 9 | 13 | … | 0 |
O | 10 | 31 | … | 2 |
… | … | … | … | … |
NH3 | 3 | 9 | … | 0 |
Далее двумерный массив 15х15 преобразовывался в одномерный, вида 120х1 (табл. 2).
Таблица 2 - Матрица атомов RBD-ACE2 для структуры 6LZG, расположенных на расстоянии 6-10Å
NH1 vs NH1 | NH1 vs O | O vs O | NH3 vs NH1 | NH3 vs O | NH3 vs NH3 | .. (n =120) | |
6LZG | 9 | 23 | 31 | 3 | 11 | 0 | .. |
Для каждого из выбранных интервалов расстояний составлены матрицы, таких интервалов 4, следовательно, количество признаков для одного комплекса составляет 4×120 = 480 признаков.
На полученных вышеприведенных матрицах произведена проверка соответствия структурных комплексов, посредством критерия Брея-Кертиса в пакете программ scipy. Несходство Брея-Кертиса измеряется в диапазоне от 0 до 1, где 0 означает, что два образца имеют одинаковый состав (то есть они имеют общие количественные признаки), а 1 означает, что два образца не имеют общих признаков. Стоял вопрос о возможном исключении комплексов с низким разрешением из-за неопределенности координат атомов в структурах, определенных криоэлектронной спектроскопией. По итогу, комплексы с низким разрешением (>3 Å) были также включены в рассмотрение, поскольку критерий не превышал для всей выборки значение 0,2, что говорит о высокой структурной согласованности комплексов.
3. Результаты и обсуждение
При подробном изучении межатомных расстояний в исследуемых структурах был выделен ряд особенностей. Так, минимальное среди всех структур расстояние между атомами N (2,5 Å) наблюдается для GLN42 (NE2, NH2) – GLN498 (NE2, NH2) в структуре 7EKE, и 3,0 Å в структурах 7E3J, 7VX4. Во всех этих случаях связей эти атомы N между собой не образуют. Но всё было бы иначе в случае разворота остатка Q498: образовались бы водородные связи сразу и с Q42, и с Y501. По-видимому, это говорит о том, что конформация Q498 в этих структурах не является оптимальной.
В целом ситуация близкого расположение двух атомов азота, являющихся донорами атомов водорода, довольно распространена: GLN42 (NE2, NH2) – GLN498 (NE2, NH2) и LYS353 (NZ, NH3) – ASN501 (ND2, NH2) в 7VX5; GLU35 (N, NH1) – GLN493 (NE2, NH2) в 7EKE. Пары с бóльшим расстоянием между атомами взаимодействия также не демонстрируют и, на первый взгляд, особого интереса не представляют.
Вместе с тем контакты HIS34 (NE2, NR2, NY) – GLN493 (NE2, NH2) на расстоянии 3 Å в структурах 7SY2, 7SY6, 7SY8 и HIS34 (NE2, NR2, NY) – LYS417 (NZ, NH3) на расстоянии 3,3 Å в структуре 7KJ2 являются примером редкой в белках водородной связи между атомами азота
.На редкость минимальное среди всех структур расстояние между атомами O (2,2–2,3 Å) наблюдается и в случае атомов TYR83 (OH, OH1) – ASN487 (OD1, O) 7VX4 и 7VX5. Напротив, эта же пара в большинстве других структур находится на значительно большем расстоянии (3–4 Å). Такое нетипичное расстояние между атомами, наблюдаемое в структурах, полученных с низким разрешением (3,8–3,9 Å), возможно, говорит о недостаточном качестве конформации в последних.
Между тем, выявлено несколько признаков, которые могут быть интерпретированы, как образование связи между остатками посредством одной или большего числа молекул воды. В частности, таковой является связь между остатками G354 (ACE2) и V503 (RBD), опосредованная двумя молекулами воды (наблюдается в структуре 6LZG и ряде других из PDB).
Далее, на основании полученных матриц, проводился сравнительный анализ групп комплексов с разными показателями аффинности.
Для выявления структурных паттернов, для заданных групп комплексов использован U-критерий Манна–Уитни, такой подход позволил выделить 34 статистически-значимых (p-value <0,05) дифференциальных признака из 480. На рисунке 3 на тепловой карте изображены наиболее значимые 22 из них.
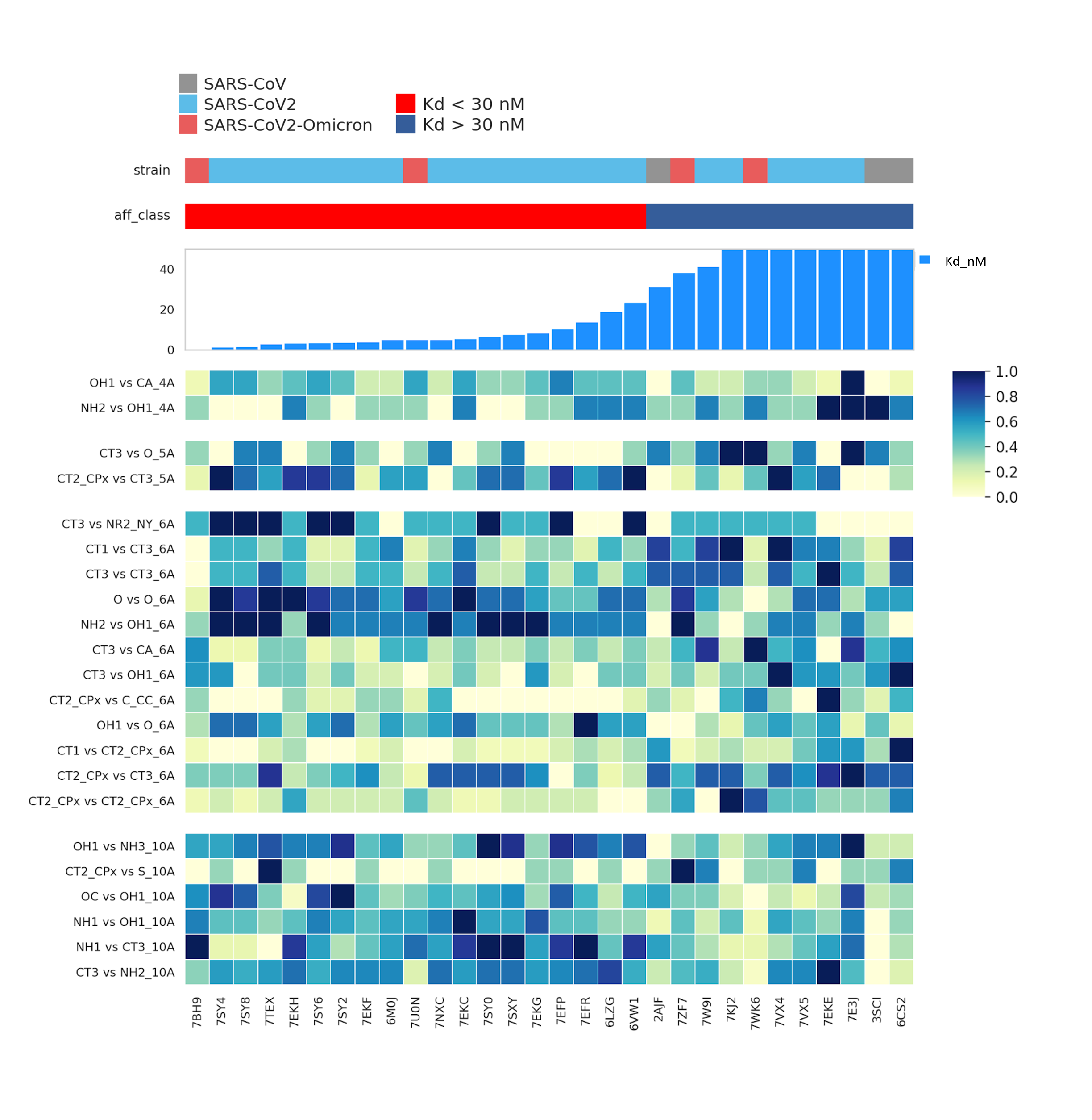
Рисунок 3 - Репрезентация структурных паттернов, отличающихся среди двух групп аффинности
Примечание: отражены 22 наиболее значимых признака выявленных U-критерием, 4 блока тепловой карты - 4 диапазона дистанций, префикс к каждому признаку обозначает крайнюю границу диапазона (4А = 2-4Å, 5А = 4-5Å, 6А = 5-6Å, 10А = 6-10Å), абсолютные значения нормированы медианным шкалированием внутри каждого признака для удобства визуального восприятия
Несмотря на значительное число полярных атомов на интерфейсе, не было выявлено признаков, которые можно было бы интерпретировать как образование H-связи непосредственно между остатками. Это означает, что высоко- и низкоаффинные комплексы не отличаются друг от друга по количеству H-связей между атомами белка.
На рисунке 4 подробнее рассмотрены аминокислотные пары для контакта OH1-NH2 на расстоянии 5-6 Å, количество которого положительно влияет на аффинность связывания (p-value<0,01). Так, можно выделить две значимые пары, имеющие данный контакт, представленные в основном в высокоаффинных комплексах: GLN42-TYR449 и GLN498-TYR41. Как уже было выявлено ранее GLN42 ACE2 и TYR449 RBD действительно образуют водородную связь и участвуют в стабилизации hot spot 353 [16], а пара GLN498(RBD)-TYR41(ACE2) образует Ван-дер-Ваальсовы взаимодействия и стабилизирует эту же область связывания на интерфейсе
.
Рисунок 4 - Наиболее значимые аминокислотные пары, образующие контакт NH2-OH1 на расстоянии 5-6 Å
Рисунок 5 - Наиболее значимые аминокислотные пары, образующие контакт NH3-OH1 на расстоянии 4-5 Å
По итогу для всех значимых признаков производилось определение конкретных а/о (аминокислотных остатков), атомы которых входят в тот или иной признак. На рисунке 6 приведены наиболее характерные пары а/о и разница частот их встречаемости для всех отобранных контактов. Можно заметить, что в зависимости от пары K353 (ACE2) кардинально может измениться аффинность комплекса. Варианты Y505 и Y501 (RBD) являются характерными для группы комплексов высокой аффинности; варианты R403, Y484, Y491 (RBD) - характерны для группы комплексов низкой аффинности.

Рисунок 6 - Разница частоты встречаемости значимых а/о (аминокислотных остатков), с нормировкой на размер выборок, применительно к группам низкоаффинных и высокоаффинных комплексов
Примечание: в отрицательных значениях - пары а/о, характерные для низкоаффинных комплексов, в значениях больше 10 - характерные для высоко-аффинных комплексов, центральная группа от 0 до 10 - контакты характерные для двух групп
4. Заключение
Полученные результаты открывают перспективу использования данных о межатомных расстояниях для оценки аффинности связывания не только между RBD и ACE2, но и в других белок-белковых комплексах. Кроме того, большое число выявленных статистически значимых признаков, относящихся к неполярным атомам, свидетельствуют о важности гидрофобных контактов и расположения относительно них молекул воды в образовании комплексов RBD-ACE2.