Молекулярный докинг в медицине. Обзор литературы
Молекулярный докинг в медицине. Обзор литературы

Аннотация
Молекулярный докинг — это метод компьютерного моделирования, предназначенный для предсказания предпочтительной ориентации одной молекулы относительно другой при образовании ими стабильного комплекса., что особенно важно при разработке лекарственных средств. В данной работе целью является проведение всестороннего обзора литературы, посвященного основным принципам молекулярного докинга, используемому программному обеспечению и его роли в процессе создания лекарственных препаратов. Методологическая часть исследования основана на поиске и анализе публикаций в ведущих базах данных, таких как Scopus, PubMed, Web of Science, Google Scholar и SpringerLink, с использованием ключевых слов, связанных с молекулярным докингом и разработкой лекарств.
В результате исследования проанализированы основные преимущества и ограничения различных программных пакетов для молекулярного докинга, а также освещены современные тенденции в применении машинного обучения и искусственного интеллекта для повышения точности виртуального скрининга. Таким образом, данное исследование подчеркивает важность молекулярного докинга как ключевого инструмента в разработке новых лекарств, а также его перспективы в сочетании с новейшими технологиями для увеличения эффективности поиска потенциальных лекарственных соединений.
1. Введение
Молекулярный докинг – это метод компьютерного моделирования, который предсказывает предпочтительную ориентацию одной молекулы по отношению к другой при образовании ими стабильного комплекса, а также энергию и режим связывания полученного комплекса. Этот метод играет значительную роль в открытии и разработке лекарств, позволяя моделировать взаимодействия между малыми молекулами и их биологическими мишенями, такими как белки или нуклеиновые кислоты. Моделируя эти взаимодействия, докинг позволяет идентифицировать потенциальных кандидатов на роль новых лекарственных средств, изучать механизмы их действия на молекулярном уровне и оптимизировать их структуру для достижения желаемой аффинности и специфичности , .
С развитием вычислительных технологий молекулярный докинг стал более точным и доступным для широкого круга исследователей. На протяжении десятилетий метод претерпел значительные изменения: от подходов, рассматривающих лиганд и рецептор как жесткие структуры, до современных подходов, способных учитывать гибкость рецептора, и подходов, интегрированных с искусственным интеллектом и машинным обучением , . Метод молекулярного докинга активно используется в совокупности с методом молекулярной динамики, позволяющей подтвердить результаты докинга in silico, оптимизировать структуры полученных комплексов, а также предоставить более углубленное понимание механизма связывания рецептора с лигандом , .
В настоящее время, молекулярный докинг является неотъемлемым инструментом структурной биологии и медицинской химии для решения широкого спектра задач. Поскольку спрос на новые терапевтические средства продолжает расти, молекулярный докинг позволяет повысить эффективность процесса открытия лекарств за счет проведения высокопроизводительного скрининга обширных библиотек химических соединений, сокращая затраты времени и ресурсов
. Кроме того, молекулярный докинг способствует обнаружению диагностических маркеров и изучению фундаментальных биохимических процессов, а также может быть интегрирован в персонализированную медицину, позволяя разрабатывать лекарства, адаптированные к индивидуальным генетическим профилям пациентов , , . Таким образом, молекулярный докинг представляет собой мощный инструмент для решения различных проблем современного здравоохранения.Цель данного обзора — провести комплексный анализ молекулярного докинга, охватывающий его основные принципы, используемое программное обеспечение, роль в разработке лекарств, а также обсудить перспективы его применения и существующие вызовы.
2. Молекулярный докинг
Молекулярный докинг – это метод компьютерного моделирования, который используется для предсказания взаимодействий между двумя молекулами, где одна из них выступает в роли рецептора, а другая – лиганда. Схема молекулярного докинга представлена на рисунке 1. Рецепторами могут быть белки, нуклеиновые кислоты или другие биомолекулы, однако большинство программ для молекулярного докинга оптимизировано для работы с белками . Лигандами обычно являются малые молекулы, но в случае белок-белкового докинга лигандом может быть другой белок. Основная цель молекулярного докинга — предсказать, как лиганд связывается с активным участком рецептора, соответствующие физико-химические молекулярные взаимодействия, а также оценить аффинность (силу связывания) и стабильность образовавшегося комплекса.
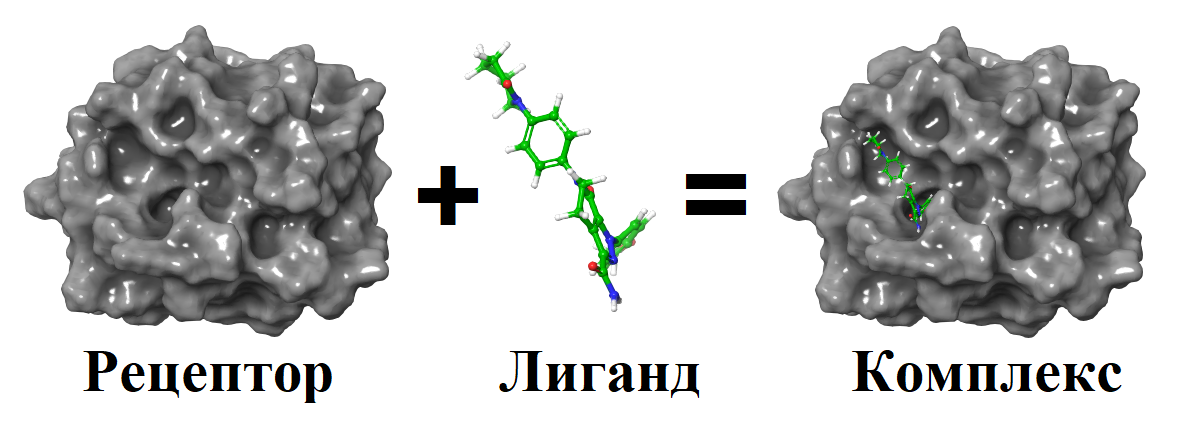
Рисунок 1 - Схема молекулярного докинга
Структуры лигандов могут быть получены из баз данных малых молекул, таких как PubChem и ZINC . Если трехмерная структура лиганда отсутствует в базах данных, она может быть смоделирована на основе двухмерной структуры с использованием программ, таких как ChemSketch, MarvinSketch, ChemDraw и др. .
После подготовки структур необходимо определить сайт связывания (связывающий карман) на рецепторе, куда будет осуществляться молекулярный докинг лиганда. Чаще всего информация о сайте связывания известна и доступна в литературе или базах данных. Однако в отсутствие такой информации используются алгоритмы для предсказания потенциальных сайтов связывания или проводится «слепой» молекулярный докинг, охватывающий весь рецептор, который требует значительных вычислительных ресурсов .
Во время расчетов молекулярного докинга общепринятой стратегией является использование сеточных полей (гридов), представляющих собой трехмерную сетку, которая накладывается на рецептор или на его связывающий карман, чтобы определить потенциальные точки взаимодействия между молекулами. Это помогает сократить время расчетов, ограничивая область оценки энергетических взаимодействий лиганда с рецептором .
Следующий этап включает генерацию различных конформаций и ориентаций (поз) лиганда в пределах сайта связывания рецептора. Это достигается путем исследования вращающихся связей лиганда, принимая во внимание его рото-трансляционные и внутренние степени свободы, а также перемещения лиганда в пределах сайта связывания . Следует отметить, что большое количество вращающихся связей увеличивает степени свободы лиганда, тем самым увеличивая количество возможных конформаций, что сказывается на вычислительных затратах .
После генерации возможных поз лиганда проводится оценка и ранжирование комплексов лиганд-рецептор с использованием оценочных функций . Схема принципа действия оценочных функций представлена на рисунке 2. Цель оценочной функции – отделить правильные позы от неправильных за разумное время вычислений. Для каждого возможного комплекса лиганд-рецептор программное обеспечение вычисляет потенциальную энергию связывания, которая отражает пространственное и химическое соответствие лиганда связывающему карману рецептора. Полученные комплексы с самой низкой энергией связывания рассматриваются как наиболее вероятные кандидаты для использования в дальнейших исследованиях.
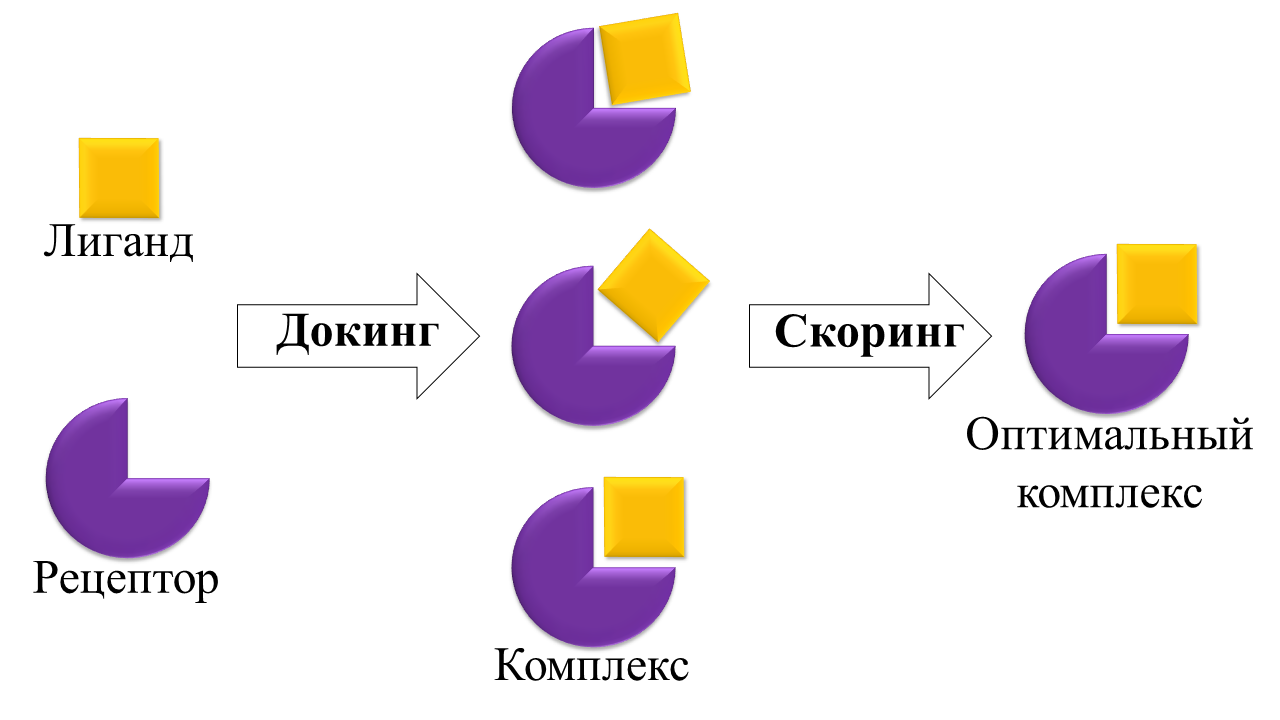
Рисунок 2 - Схема принципа действия оценочной функции, где скоринг – это процесс оценки и ранжирования комплексов лиганд-рецептор
При проведении молекулярного докинга лиганд и рецептор могут рассматриваться как жесткие и как гибкие структуры. Под «гибкостью» подразумевается способность молекул, участвующих в докинге, изменять свою конформацию во время процесса связывания . Так, жесткий докинг не учитывает конформационные изменения молекул при их взаимодействии и учитываются только три трансляционные и три вращательные степени свободы, в связи с чем жесткий докинг является более быстрым, но менее точным подходом. Гибкий докинг, в свою очередь, позволяет учитывать конформационные изменения лиганда и рецептора, однако данный подход требует высоких вычислительных затрат и может сталкиваться с плохим моделированием гибкости рецептора , . Компромиссом между точностью и временем вычислений, является подход полугибкого докинга, рассматривающего лиганд как гибкий, в то время как рецептор остается жестким . Существует также ансамблевый докинг – альтернативная стратегия, при которой используется набор конформаций рецептора с различными связанными лигандами, обычно экспериментально определенными, для каждой из которых проводится жесткий докинг с последующим объединением всех результатов , .
Таким образом, молекулярный докинг представляет собой мощный инструмент для предсказания взаимодействий между молекулами и позволяет оценивать аффинность и стабильность образованных комплексов. Более того, метод может использоваться для определения наиболее правдоподобной конформации связывания лиганда с рецептором и улучшения показателей связывания, путем точечных модификаций лиганда или мутаций определенных аминокислотных или нуклеотидных остатков рецептора. Несмотря на то, что подходы молекулярного докинга могут быть ресурсоемкими в плане затрачиваемого времени и требуемых вычислительных мощностей, они значительно дешевле и быстрее, а также более доступны, чем соответствующие экспериментальные подходы. Однако вычислительные подходы предоставляют только гипотетические предсказания поведения системы, которые требуют экспериментального подтверждения , . Поэтому наиболее разумным подходом для исследования сложных биологических и фармакологических систем является комбинирование вычислительных и экспериментальных подходов.
3. Программное обеспечение для молекулярного докинга
За последние десятилетия было разработано более 60 различных инструментов и программ для молекулярного докинга для академического и коммерческого использования . Каждая из этих программ имеет свои уникальные алгоритмы, методы оценки и возможности для обработки данных. Эти программы могут варьироваться от простых инструментов для быстрых вычислений до комплексных платформ, предлагающих гибкие настройки и возможность проведения высокоточных расчетов. В таблице 1 представлены некоторые известные программы молекулярного докинга и их описание.
Таблица 1 - Известное программное обеспечение для молекулярного докинга
Программное обеспечение | Описание | Преимущества | Ограничения | Ссылка |
AutoDock4 | Автоматизированный инструмент для молекулярного докинга. Состоит из двух основных программ: autodock выполняет докинг лиганда с набором гридов, описывающих рецептор; autogrid предварительно рассчитывает эти гриды. | Гибкость рецептора; «слепой» молекулярный докинг; оценочная функция на основе линейного регрессионного анализа; силовое поле AMBER; большой набор комплексов белок-лиганд с известными константами ингибирования; хорошая корреляция между предсказанными константами ингибирования и экспериментальными данными. | Часто приводит к ненадежным результатам при докинге с гибкими сайтами связывания; не может учитывать гибкость циклических и макроциклических лигандов. | |
AutoDock Vina | Программное обеспечение для молекулярного докинга; является компонентом в AutoDock Suite, вместе с AutoDock4. | Работает на несколько порядков быстрее AutoDock4; может использовать несколько процессоров или ядер, что ускоряет работу; некоторые боковые цепи рецептора можно выбрать так, чтобы они рассматривались как гибкие. | В первую очередь использует жесткую модель рецептора, игнорируя динамическую структурную гибкость белка; опирается на неполяризуемые силовые поля; неполное представление ароматических π-π взаимодействий | |
DockingServer | Веб-интерфейс, который для молекулярного докинга. | Удобный веб-интерфейс; полный контроль над настройкой параметров. | Файлы выходной структуры большие, а пространство для хранения ограничено. | |
SwissDock | Веб-сервис для молекулярного докинга, который использует два алгоритма: притягивающие полости и AutoDock Vina. | Удобный интерфейс; автоматизированная подготовка лиганда и рецептора; возможность проведения «слепого» докинга | Докинг только индивидуального лиганда. | |
PyRx | Программное обеспечение для виртуального скрининга, объединяющее несколько программ с открытым исходным кодом, включая AutoDock и AutoDock Vina. | Простой и функциональный пользовательский интерфейс; запуск с любой платформы. | Бесплатная версия – это старая версия, которая больше не поддерживается и имеет ограниченный функционал. | |
FlexAID | Алгоритм гибкого докинга с оценочной функцией, основанной на поверхностной комплементарности. | Гибкий докинг; не требуется ручная настройка структуры рецептора. | Необходимо отдельно установить программу для визуализации; могут возникать ошибки при наличии в структуре рецептора редких типов атомов. | |
LeDock | Кроссплатформенная программа для быстрого и точного гибкого докинга, использует комбинацию генетического алгоритма и имитации отжига. | Быстрый гибкий докинг; высокая точность; идентификация поз связывания, близких к нативным; удобный интерфейс; кроссплатформенность. | Нужно дополнительное программное обеспечение для подготовки рецептора и визуализации; невозможно изменить количество поз лиганда. | |
Glide | Комплексное программное обеспечение для молекулярного докинга и виртуального скрининга. Использует две различные оценочные функции, SP и XP GlideScore. | Эффективный скрининг больших библиотек; простота и доступность использования; удобный графический интерфейс; высокая точность; множество настраиваемых параметров; гибкий докинг; может учитывать молекулы воды в активном сайте рецептора; имеет возможность одновременного расчета нескольких заданий; кроссплатформенность. | Не всегда может адекватно моделировать связывание ионов металлов из-за ограничений в силовых полях; ограниченная способность учитывать динамическую гибкость лигандов и рецепторов; для каждого программного модуля требуется отдельная лицензия. | |
GOLD (генетическая оптимизация для докинга лигандов) | Программное обеспечение для молекулярного докинга на основе генетического алгоритма, которое использует адаптивные функции оценки. | Гибкий докинг; определяемые пользователем функции оценки; множество вариантов настроек с включением молекул воды и атомов металлов; оптимизирован для параллельного выполнения в процессорных сетях; позволяет атомное перекрытие между белком и лигандом; кроссплатформенность. | Требует определения отчетливого вогнутого сайта связывания, в связи с чем не подходит для белок-белкового докинга. | |
Molecular Operating Environment (MOE) | Комплексный инструмент для компьютерной разработки лекарств, который объединяет визуализацию, моделирование и симуляции, а также разработку методологии в одном программном обеспечении. | Настраиваемый доступный исходный код; кроссплатформенность; удобный графический интерфейс; показывает взаимодействующие аминокислоты с положением; многопрофильность приложений и функций; предлагает поддержку для других инструментов молекулярного докинга. | Неожиданные результаты с его модулем подготовки белка, т.к. в некоторых случаях он выдает неправильные заряды. | |
rDock | Программное обеспечение с открытым исходным кодом на базе Linux для молекулярного докинга; в первую очередь предназначено для высокопроизводительного виртуального скрининга и прогнозирования режима связывания. | Высокая скорость вычислений; широкий спектр внешних настроек; возможность имитации гибкости рецептора; может быть установлен на вычислительном кластере и развернут на неограниченном количестве центральных процессоров. | Требуется привязка OpenBabel Python; крутая кривая обучения; правила, используемые для протонирования и депротонирования, довольно грубы и не настраиваются пользователем; может доставить проблемы, если пользователь никогда не компилировал из исходников. | |
OEDocking | Набор инструментов и рабочих процессов молекулярного докинга, каждый из которых разработан для решения различных аспектов взаимодействия белок-лиганд. | Удобный графический интерфейс; систематическое и нестохастическое исследование всех возможных поз белок-лиганд, фильтров для комплементарности формы и выравнивания химических признаков перед выбором и оптимизацией поз; высокая скорость вычислений; возможность учитывания гибкости рецептора. | Может возникнуть проблема с эффективным отбором высокогибких лигандов или конформаций рецепторов, что может привести к пропуску режимов связывания или неточным прогнозам в системах со значительной конформационной изменчивостью. |
Данные, представленные в таблице 1, демонстрируют разнообразие инструментов для молекулярного докинга, каждый из которых обладает уникальными характеристиками, направленными на решение различных задач в области биоинформатики и структурной биологии. Программы отличаются как по своей функциональности, так и по специфике применения, предлагая пользователю различные подходы к молекулярному моделированию и виртуальному скринингу. Так, одни программы ориентированы на более точный и гибкий докинг, обеспечивая возможность настройки множества параметров и поддержку сложных вычислительных задач, в то время как другие предлагают более простые и интуитивно понятные интерфейсы, что облегчает их использование для менее опытных пользователей. При этом веб-сервисы для молекулярного докинга, например, SwissDock, предоставляют удобный доступ через интернет и автоматизированные настройки, что может быть особенно полезно для пользователей, предпочитающих простой и быстрый запуск расчетов без необходимости глубокой настройки параметров. В то же время коммерческие программы, такие как Glide, GOLD и MOE, имеют высокую стоимость, но являются комплексным программным обеспечением с широким набором функций и удобным интерфейсом. Бесплатное программное обеспечение с открытым исходным кодом, такое как rDock и AutoDock, также позволяет провести качественный молекулярный докинг и может предоставить большую гибкость и возможность кастомизации, но при этом может требовать сложной настройки и использования дополнительных инструментов и модулей.
Таким образом, представленные программы обладают достаточным функционалом для проведения успешного докинга, но их эффективность и удобство зависят от конкретных задач и условий, в которых они используются. В конечном итоге выбор подходящего программного обеспечения для молекулярного докинга определяется спецификой задачи, точностью и производительностью программы, возможностью интеграции с другими инструментами, качеством поддержки и документации, а также доступностью ресурсов и стоимостью самого программного обеспечения.
4. Применение молекулярного докинга в разработке лекарственных средств
Открытие и разработка лекарственных средств является очень длительным и трудоемким процессом, для оптимизации которого в последнее время активно применяются in silico методы, в том числе молекулярный докинг. Молекулярный докинг позволяет идентифицировать новые лекарственные соединения, на молекулярном уровне предсказывать взаимодействия лиганда и мишени, а также определять взаимосвязи структура-активность. В настоящее время докинг является одним из самых успешных подходов в компьютерном проектировании лекарственных средств, позволяя проводить виртуальный скрининг больших библиотек химических соединений за доступное время, тем самым снижая затраты на идентификацию перспективных соединений и повышая шансы нахождения желаемых кандидатов в лекарственные средства , . Так, на основе молекулярного докинга разработаны крупномасштабные протоколы виртуального скрининга для идентификации новых рецепторов для известных лигандов, связывающих карманов белков, лигандов, активных против заданной мишени или набора мишеней, а также нежелательных побочных эффектов лекарственных средств , , . Молекулярный докинг может использоваться как самостоятельный метод, однако на практике его часто интегрируют в рабочие процессы, включающие другие вычислительные и экспериментальные подходы , .
Стратегии компьютерного проектирования лекарств можно разделить на два типа: разработка лекарств на основе лигандов и разработка лекарств на основе структуры. Разработка лекарств на основе лигандов – это подход, который используется при отсутствии информации о трехмерной структуре рецепторов и основан на знании структуры молекул лигандов, связывающихся с биологической мишенью. Основными инструментами данного подхода являются трехмерные количественные соотношения «структура-активность» (3D QSAR) и моделирование фармакофоров, которые могут предоставить прогностические модели для идентификации и оптимизации перспективных соединений . QSAR основан на предположении, что структура молекулы лиганда, а именно ее электронные, стерические и геометрические свойства, соотносятся с физико-химическими свойствами молекулы, ее токсичностью и биологической активностью . Моделирование фармакофоров, в свою очередь, основано на том, что набор структурных характеристик молекулы, таких как доноры и акцепторы водородной связи, гиброфобность и наличие заряженных участков, распознается связывающим карманом рецептора и отвечает за биологическую активность этой молекулы .
Подходы на основе лигандов активно используются для выбора наиболее подходящих конформаций белков для молекулярного докинга. В ряде исследований было показано, что сочетание подходов на основе лигандов и молекулярного докинга значительно улучшает прогнозы виртуального скрининга . Например, переоценка результатов докинга с помощью дополнительной оценочной функции основе фармакофоров и использование 3D- и форма-зависимого сходства могут существенно повысить точность прогнозов , , . Однако следует отметить, что подходы на основе лигандов наиболее эффективны в случаях, когда есть хотя бы один зарегистрированный сокристаллизованный лиганд.
Разработка лекарств на основе структуры основана на использовании трехмерной структуры рецептора для разработки молекул-ингибиторов, которые могут эффективно взаимодействовать с этим рецептором. Данный подход включает разработку лекарств de novo и виртуальный скрининг, ключевым инструментом которых является молекулярный докинг. Так, основой разработки лекарств de novo является информация о составе и ориентации аминокислот, формирующих сайт связывания рецептора, которая может быть использована для разработки специфичных для этого рецептора лигандов. Виртуальный скрининг, в свою очередь, используется на этапе идентификации перспективных соединений и заключается в молекулярном докинге большой библиотеки соединений с сайтом связывания рецептора, с последующей оценкой и ранжированием полученных комплексов . Далее наиболее перспективные соединения подвергаются экспериментальному анализу для выявления и подтверждения желаемой биологической активности.
Для улучшения результатов виртуального скрининга молекулярный докинг широко используется в сочетании с другими подходами на основе структуры, такими как молекулярная динамика и оценка свободной энергии связывания. В частности, молекулярная динамика позволяет оценить гибкость аминокислотных остатков в связывающем кармане рецептора и стабильность предсказанных комплексов, а также исследовать крупные конформационные изменения данного рецептора , . Особенно перспективно использование молекулярной динамики в отношении гибких рецепторов с ограниченным количеством зарегистрированных кристаллографических конформаций. Например, Wang и соавт. провели моделирование молекулярной динамики для оценки стабильности структуры β-амилоида 42, в результате которого была определена репрезентативная конформация белка для проведения виртуального скрининга библиотеки коммерчески доступных соединений.
В компьютерной разработке лекарств молекулярный докинг может использоваться и для множества других задач. Например, обратный молекулярный докинг позволяет предсказывать биологические мишени для целевого лиганда путем обратного скрининга лиганда в библиотеке структур белков , . Для проведения обратного скрининга лиганда требуется использование подходящих баз данных или библиотек мишеней, среди которых одной из самых известных является PDTD, содержащая информацию о структурах белков, заболеваниях, биологических функциях и лекарствах . Обратный докинг может быть полезным инструментом для выявления побочных эффектов лекарственных средств, а также позволяет прогнозировать нежелательные взаимодействия с нецелевыми мишенями , . Однако следует отметить, что получение хороших прогнозов скрининга с использованием обратного докинга напрямую связано с качеством и полнотой данных, представленных в базе данных мишеней .
Молекулярный докинг также используется для разработки многоцелевых лигандов в рамках полифармакологии, которая фокусируется на создании химических соединений, способных одновременно воздействовать на несколько терапевтически значимых мишеней , . Однако процесс разработки многоцелевых лигандов является сложной задачей, успех которой зависит от выбора конформации и структуры сайта связывания мишени .
Перепрофилирование (перепозиционирование) лекарственных средств представляет собой важный подход к открытию новых терапевтических применений уже существующих лекарственных препаратов, натуральных соединений и соединений, находящихся на стадии клинических испытаний , . Молекулярный докинг играет ключевую роль в этом процессе, поскольку позволяет идентифицировать новые мишени для уже известных лигандов. Например, Kinnings и соавт. провели исследование структур редуктазы белка-переносчика еноил-ацила (InhA) Mycobacterium tuberculosis и выявили, что энтакапон, одобренный для лечения болезни Паркинсона, активен против резистентных штаммов Mycobacterium tuberculosis и может быть потенциально использован для лечения туберкулеза.
Молекулярный докинг также позволяет использовать предсказанные позы связывания лигандов для оптимизации аффинности связывания и модификации характеристик лиганда для достижения желаемой биологической активности. Так, предсказанные позы могут быть использованы для проектирования лигандов, которые селективно связываются с одной конкретной мишенью набора связанных мишеней ; идентификации участков структуры лиганда, которые можно модифицировать флюоресцентными или химическими метками, не нарушая связывания лиганда с рецептором ; идентификации химических модификаций структуры лиганда, которые позволяют оптимизировать различные свойства лиганда, например, токсичность или растворимость, без ухудшения аффинности связывания с рецептором ; для создания двухвалентных лигандов или лигандов, нацеленных на сайты связывания двух разных молекул . Таким образом, позы лигандов, предсказанные молекулярным докингом, позволяют изучить структурный механизм, посредством которого лиганды влияют на функцию мишени.
Молекулярный докинг активно применяется на различных этапах разработки лекарственных средств в широком спектре различных исследований, начиная от изучения перспектив использования морских соединений и нутрицевтиков для лечения различных заболеваний , , изучения взаимодействия пищевых ферментов с их субстратами , , поиска лекарства от вируса иммунодефицита человека , до использования нуклеиновых кислот в качестве мишени для лечения рака и антимикробной терапии и для многих других применений. Отдельно следует отметить применение докинга в персонализированной медицине. Молекулярный докинг позволяет делать прогнозы о том, как генетические данные пациента могут повлиять на взаимодействие лекарств с рецепторами, что потенциально повышает терапевтическую эффективность при одновременном снижении побочных эффектов и способствует разработке фармацевтических препаратов, адаптированных к уникальному генетическому профилю и молекулярному составу каждого пациента , . Таким образом, молекулярный докинг представляет собой универсальный инструмент для решения широкого спектра задач в области открытия и разработки лекарственных средств, помогая создавать более безопасные и эффективные лекарства, имеющие больше шансов на успех в клинических испытаниях.
5. Перспективы и ограничения
С момента своего появления в середине 1970-х годов молекулярный докинг стал важным и универсальным инструментом в открытии и разработке лекарственных средств. Он используется для поиска новых химических структур в крупных библиотеках соединений, полифармакологии, прогнозирования побочных эффектов и профилирования для повторного использования лекарственных препаратов. В настоящее время докинг активно интегрируют в автоматизированные рабочие процессы для скрининга больших библиотек соединений и мишеней. В этом отношении ключевую роль сыграли современные достижения в области высокопроизводительных вычислений, что позволило выполнять in silico скрининг миллионов соединений в разумные сроки . Так, благодаря применению продвинутых алгоритмов и моделей, таких как нейронные сети и глубокое обучение, можно эффективно анализировать большие объемы данных и повышать точность и скорость прогнозирования за счет моделей, обученных на обширных наборах данных комплексов белок-лиганд , , . Использование современных графических процессоров также сделало анализ больших данных доступным для более широкой аудитории, и ожидается, что они сыграют ключевую роль не только в докинге, но и для компьютерного проектирования лекарств в целом .
Учитывая потенциал молекулярного докинга, ведется активная работа по преодолению его внутренних ограничений. Так, дальнейшего усовершенствования требуют оценочные функции, которые часто дают результаты, не соответствующие экспериментальным аффинностям связывания . Существуют успешные примеры, показавшие, что с помощью докинга возможно идентифицировать перспективные соединения из огромной базы данных и разрабатывать на их основе лекарственные препараты. Однако реалистичность взаимодействий между лигандами и рецепторами по-прежнему требует подтверждения с помощью экспериментальных методов. Разработка более точных оценочных функций, которые требуют минимальных вычислительных ресурсов для своей работы, могут вывести применение молекулярного докинга на новый уровень.
Гибкость рецептора, особенно гибкость элементов рецептора, участвующих в связывании лиганда, также является серьезным ограничением. Некоторые методы, позволяющие учитывать гибкость боковой цепи рецептора, показали свою эффективность и адекватность при определенных условиях. Что касается глобальной гибкости, ансамблевый докинг является популярным решением, но в то же время имеет высокие вычислительные затраты, и требует эффективного способа получения и выбора надежных структур белка, которые наиболее вероятно обеспечат успешное связывание с лигандом.
Использование молекулярного докинга в разработке лекарственных препаратов также ограничено теми рецепторами, для которых доступны кристаллические структуры. Для преодоления этого ограничения было предложено несколько подходов. Например, отсутствие трёхмерных структур можно компенсировать с помощью моделирования по гомологии с использованием структурных шаблонов, высоко гомологичных целевому белку. Помимо этого, благодаря успехам в структурной биологии происходит постоянное пополнение баз данных новыми кристаллическими структурами белков и комплексов лиганд-белок, полученных экспериментально .
Различные методы переоценки и уточнения результатов докинга способствуют улучшению точности виртуального скрининга и лучшему соответствию экспериментальным данным. В этом направлении активно разрабатываются различные методики виртуального скрининга, в том числе и с применением методов на основе машинного обучения , , . Использование таких комбинированных подходов также повышает точность прогнозов и позволяет более эффективно использовать данные из различных источников. Однако важно учитывать, что каждый вычислительный метод имеет свои ограничения, которые могут затруднять их совместное использование с докингом или снижать точность предсказаний. Например, подходы на основе искусственного интеллекта, основанные на существующих данных о биологической активности и химических структурах, могут оказаться менее эффективными для мишеней, которые слабо охарактеризованы. В то же время, молекулярная динамика и расчёты свободной энергии связывания могут улучшить точность результатов даже для плохо охарактеризованных мишеней, например, помогая определить конформационные состояния, которые могут быть использованы для структурного анализа, изучения конформационного пространства рецепторов, более точно оценки сродства лиганда к рецептору и описания динамического поведения полученного комплекса , . Существующее разнообразие вычислительных инструментов предоставляет множество перспектив для расширения возможностей молекулярного докинга, а постоянные улучшения в аппаратном и программном обеспечении способствуют его эффективной интеграции в рабочие процессы. Кроме того, по мере роста объёмов общедоступной структурной, химической и биологической информации, а также её реализации в веб-платформах, базах данных и автоматизированных системах, открываются новые возможности для объединения различных методов и данных. В условиях высоких показателей отсева на этапах разработки лекарственных средств, использование докинга в сочетании с обсуждаемыми здесь подходами станет ключевым фактором для сокращения затрат и времени на создание более сложных и инновационных лекарственных препаратов с улучшенными профилями безопасности и отсутствием побочных эффектов, а также для перепрофилирования уже существующих препаратов.
6. Заключение
Молекулярный докинг играет важную роль на различных этапах разработки лекарственных средств, выступая как мощный и эффективный инструмент, который значительно ускоряет и удешевляет процесс их создания. Докинг позволяет не только прогнозировать потенциальные побочные эффекты и устойчивость к лекарствам, но и перепрофилировать уже известные препараты, открывая новые направления исследований и выходя за рамки традиционных экспериментальных подходов. Развитие программного и аппаратного обеспечения, а также накопление данных о трехмерных структурах рецепторов, способствуют совершенствованию молекулярного докинга и других вычислительных методов, делая их незаменимыми в современном процессе открытия и разработки лекарств.
Растущий интерес научного сообщества подтверждается не прекращающимся ростом количества публикаций, посвященных использованию молекулярного докинга в различных областях. Этот вычислительный подход успешно применяется в структурно-ориентированной разработке лекарств и уже привел к созданию таких препаратов, как каптоприл, дорзоламид, саквинавир, ритонавир, индинавир, тирофибан, занамивир, алискирен, тупинтривир, иматиниб и многих других , . В Казахстане исследовательские коллективы также активно используют молекулярный докинг для различных целей, например, для изучения потенциальных ингибиторов тубулина, противовирусных соединений, а также для разработки новых противоопухолевых и антибактериальных препаратов , , , .
Таким образом, молекулярный докинг продолжает оставаться важнейшим инструментом в арсенале современных ученых, непрерывно развиваясь и открывая новые горизонты для исследований и способствуя разработке более эффективных и инновационных подходов к созданию новых лекарственных средств.