DISTANT JOINING: A SEQUENCE SAMPLING METHOD FOR COMPLEX PHYLOGENIES

DISTANT JOINING: A SEQUENCE SAMPLING METHOD FOR COMPLEX PHYLOGENIES
Funding
This work has been supported by the Federal Agency of Scientific Organisations [project number 0345-2016-005]
Conflict of Interest
None declared
Alexey A. Morozov1 *, Yuri P. Galachyants1
1 Cell Ultrastructure Department, Limnological Institute SB RAS. Ulan-Batorskaya st. 3, Irkutsk, Russian Federation 664033
*To whom correspondence should be addressed.
Associate editor: Giancarlo Castellano
Received on 18 August 2017, revised on 26 September 2017, accepted on 02 November 2017.
Abstract
Motivation: Massive parallel phylogenetic analyses allow to reconstruct phylogenetic trees for every gene in genome, typically using a set of potential homologues detected by similarity search against reference databases via BLAST or BLAST-like algorithms. However, given that the amount of similarity hits between query sequence and targets is often too high, it may be necessary to reduce number of sequences for downstream pelogenetic analyses.. Currently available automatic and semi-automatic methods for dataset reduction are error-prone and may depend on additional metadata, whereas reduction “by hand” is labour-intensive and becomes intractable once phylogenetic analysis of multiple genes is to be performed.
Results: We propose a distance-based algorithm, termed Distant Joining, for phylogenetic dataset reduction that does not require additional input except sequences analyzed. DJ was shown to robustly subsample a set of sequences with minimal loss of dataset divergence from large and complex sequence data sets. In the context of out study, the underlying assumptions and limitations of different subsampling approaches are discussed, and directions for selection of the subsampling method to build phylogenomic pipelines are provided.
Availability: Proof-of-concept Python implementation is available at https://github.com/SynedraAcus/sampler under the terms of CC-BY-4.0 license.
Supplementary information: Supplementary data are available at Journal of Bioinformatics and Genomics online.
Keywords: Phylogenetics, phylogenomics, dataset reduction.
Contact: morozov@lin.irk.ru
1 Introduction
The advancement of sequencing technologies in the last decade allowed evolutionary biologists to work with model and non-model species on the genomic scale. It has now become possible to reconstruct a phylogenetic tree for every single gene in the genome of interest. Such a collection of trees is called phylome, and phylomic databases for a variety of model organisms were established (Huerta-Ceras et al. 2014). Phylogenetic analysis, whether for a single gene or within the phylomic project, starts by identifying the potential homologs of the gene in question in some large reference database (typically BLAST search against NCBI nr). These homologs are then aligned and used for the phylogenetic tree reconstruction. However, it may be biologically counterproductive or computationally intractable to use all the high-scoring BLAST hits for the tree reconstruction. For well-sequenced genes there may be tens or hundreds of thousands of hits.While alignments and trees on that scale have been built (eg SSU and LSU rRNA SILVA (Quast et al. 2013) and Greengenes (DeSantis et al. 2006) databases), it requires both significant computational resources and competent specialists., On the other hand, phylomic pipeline should produce lots of trees in acceptable time with minimal human intervention. Therefore, hits of analyzed sequence to query database should be somehow filtered and the underlying sequence dataset should be subsampled prior to the downstream analyses. Basically, the goal of subsampling is to reduce a large dataset to a smaller representative subset. From phylogenetic point of view, “original” and “subsampled” trees sould be generally consistent and containsimilar sets of clades . This formulation of a problem does not take into account the biological meaning of a clade. Whether studying the relationships of taxa or protein families, one need at least a single sequence representing each clade. Several subsampling strategies were developed to date. Using smaller database for similarity searches, one can search only a few proteomes instead of the entire nr database or its analogue, thus ending up with several tens of hits with high e-values. This approach is used by phylomeDB (Huerta-Ceras et al. 2012) and in other studies limited to evolution of relatively small groups of species, e.g. genus Rickettsia (Murray et al. 2016). It certainly solves the problem of having too much input data, but at the cost of significantly reduced taxonomic coverage. The resulting trees will contain only the taxa whose proteomes were used initially, missing any homologs from outside this arbitrary group. HGT cases from external organisms may be detected, but clarification of the sequence origin will require additional effort; HGTs from the group to external organisms will not be detected. Monophyly of the group will not be tested, and paralogy assessment will be limited to only this group's evolutionary history. If the search is performed against the entire nr database, the dataset size can still be limited by taking a limited number of sequences with the highest similarity. This strategy can potentially result in bloating clades containing the query sequence, while the rest of the tree remains underrepresented or missing altogether. As a somewhat extreme example, consider BLAST search of A. thaliana RuBisCO large subunit (NR_051067.1) against NCBI nr. As of 2015, 71 of 100 closest hits (by identity) are eudicots, 58 of which are family Brassicaceae. This problem may not be so pronounced in other cases, but the very idea of distance-based phylogenetics is that the more similar sequences are, the more likely they are to be closely related. Thus sampling most similar sequences is, by definition, sampling most closely related sequences. If the aim of the work is to study as diverse a collection of the gene's relatives as possible, this approach is counterproductive. The third approach is to select sequences manually. If the evolutionary history of the gene in question is relatively well-studied, it is possible to just take representatives of all previously described major groups and add the novel sequences that need to be placed. This way the dataset will be as good as possible given current knowledge on the matter, but at the cost of time spent on manual data curation. It can and should be done if one studies a single gene or a group of genes, but a phylome-scale analyses require reconstructing thousands of phylogenetic trees, so it's infeasible to have an actual human working on every dataset. In addition, prior knowledge may not be available at all. A significant fraction of genes even in human genome has unknown function: only 34 thousand proteins out of almost 70 thousand sequences in human proteome at UniprotKB (The UniProt Consortium 2015; proteome up000005640) have at least one “molecular function” GO term assigned to them. The portion of unstudied genes is even higher in nonmodel organisms, and their evolutionary histories have only been addressed in phylomic projects. A recent paper (Zhou et al. 2014) proposes a novel approach to sequence sampling, dubbed AST: Automated sequence-Sampling method for improving Taxonomic divergence of phylogenetic trees. The algorithm is designed in such a way that all taxa of the same rank will be represented by approximately equal number of sequences. It was shown to outperform random sampling and similarity sampling on a series of both real and simulated datasets and come close to manual sampling where the latter is available. However, its selection procedure is based on a taxonomy and thus makes an implicit assumption that taxonomy adequately describes the evolutionary history of the sequences in question. If the gene tree is, in fact, incompatible to taxa tree, the sampling will be performed according to the latter one, potentially disregarding entire paralog families or HGT descendant groups because sequences from the same organism have been already added to the dataset. AST also uses similarity to the query sequence to select sequences within lowest-level taxa. This approach only enhances the problem, giving increased weight to the homologs of the query and undersampling its paralogs, which further hampers the algorithm performance on paralogy-heavy datasets. As discussed above, existing automated sampling methods are prone to produce misguiding datasets in complex cases. There is a need for an algorithm that would be capable to correctly sample from an arbitrary dataset. It should not depend on any data that may not be available for some of the sequences. Unclassified sequences lacking source organism or genes of unknown function with no domains predicted should not be treated worse than wellstudied genes. We have devised an automated taxonomyindependent distance-based sequence sampling algorithm that fulfills these requirements, dubbed Distant Joining, and developed a proof-of-concept implementation in Python.
2 System and Methods
The performance of distant joining algorithm and other sampling approaches was tested on two real datasets. For each of them we have defined a number of biologically significant clades and tested the percentage of those clades retained at various sampling rates. Distant Joining, AST, similarity sampling and random sampling were tested; average result of 100 replicates is reported for the latter. The first dataset included all the non-redundant nonunclassified eukaryotic SSU rRNA sequences with sequence quality and alignment quality above 90 from SILVA database release 119 (Quast et al. 2013). The clades in this cases are genera according to SILVA taxonomy. There are 4334 sequences of 2256 genera. Distance matrix was built by EMBOSS release 6.6 distmat utility (Rice et al. 2000) based on SILVA alignment using Kimura 2- parameter model. Sequence AF110418.1.2904 was used as a query for SS and AST. The second is a collection of eukaryotic chitin synthases from our earlier work (Morozov, Likhoshway 2016). It includes 137 sequences that belong to 17 groups (a mixture of eukaryotic taxa and paralogous families). Distance matrix was built using Sampler (see Implementation). Sequence of Synedra acus Chs was used as a query for SS and AST. We have also tested three out of four methods on simulated datasets. AST was excluded from this analysis because simulated data do not have an associated taxonomy. 500 trees were generated, each consisting of a number of monophyletic clades. The number of clades was taken from a normal distribution with average 15 and standard deviation of 5. Number of sequences in each clade was taken from the normal distribution as well: the clade was either large (probability 20%; avg 100, stddev 20) or small (probability 80%; avg 2, stddev 1), but all clades were set to contain at least 1 sequence. DNA sequences 200 bp long were evolved along these trees using Pyvolve (Spielman and Wilke 2015) and sampled at rates from 0.1 to 0.9 similarly to the SSU dataset. For SS, a random sequence was taken as a query. Calculations were performed on the HPC cluster of Irkutsk Scientific Center “Academician V.M.Matrosov” (https://hpc.icc.ru).
3 Algorithm and Implementation
Distant joining algorithm was named due to its slight similarity to Neighbor Joining (Saitou, Nei 1987). While the latter iteratively joins the closest pairs of sequences, DJ works by trying to find the sequence most different from those already sampled on every step. It takes distance matrix and the amount of sequences to retain as an input, and initializes subsampling set by placing a single randomly chosen sequence into it. Then it adds the sequence with the highest distance to the first one to the subsampling set (ties are resolved at random). The third sequence is chosen so that it has the highest minimal distance to those already sampled, and so on until the sample reaches the necessary size. Python-style pseudocode is shown below.

The algorithm was implemented as a Python 3.4 script. Current implementation is using EMBOSS needle for pairwise alignment and Scoredist distance estimator (Sonnhammer, Hollich 2005) for aminoacid sequences, if distance matrix is not supplied by the user.
4 Results and Discussion
Since the goal of our study was to develop the automated method that demonstrates decent performance in various complex cases, we have excluded manual selection and approaches that require preexisting data such as domain structure, GO annotation or guide trees. Four methods were compared: similarity sampling (SS), random sampling (RS), AST and DJ. They were evaluated on two drastically different real datasets: a collection of high-quality eukaryotic SSU sequences and the chitin synthase dataset from our previous work. We have also tested all methods except AST on simulated data. SSU dataset (Fig. 1a) is alarge collection of sequences with high similarity, but it is basically simple: no paralogs or HGT cases are present. As rRNA is one of the most common markers for phylogenetic, metagenomic and population genetics analyses, a plenty of high-quality fulllength sequences with reliable annotation are available from SILVA database. The only complication of the analysis is the fact that we have selected the genera as clades of interest. There are, on average, less than two sequences per genus. On the other hand, the modern diversity of chitin synthases (Fig. 1b) has been formed by a complex collection of different evolutionary events. The tree topology is influenced by multiple gene losses and duplications, the latter sometimes accompanied by domain shuffling. At least two HGT events are known to have happened. There are 17 well-supported monophyletic gene groups of various nature: some of them are taxonomically consistent (e.g. all metazoan sequences form a single clade), while some others are not (Morozov, Likhoshway 2015). For instance, fungal chitin synthases are divided into two major clades (which are further subdivided) on the distant branches of the tree. However extant fungal genomes typically contain genes from most classes of chitin synthases. The complex topology of the tree is somewhat compensated by the fact that this dataset is not as dense as the SSU rRNA one. Clades contain 4-15 sequences, and between-clade divergenceis typically higher than withinclade.
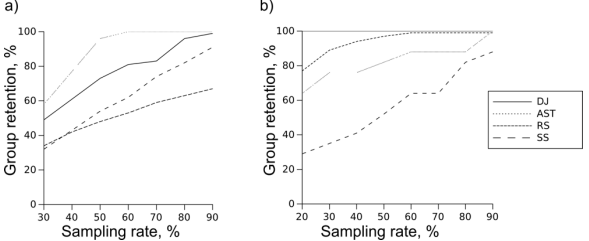
Fig. 1 -- Percentage of clades retained by different methods. a) SSU, b) Chs
5 Performance of various methods on real datasets
Percentage of clades retained by all methods at sampling rates from 20 to 90 percent is shown at Fig. 1. Random choice performs best on chitin synthase dataset, but lags behind other methods on SSU. This difference is not surprising: the more sequences there are in the clade, the more likely at least one of them will be included in the reduced set. The opposite is also true: small clades tend to get missed by random sampling. When Chs dataset is analyzed, there is a plenty of sequences in every clade, so even at the lowest sampling rates most of the clades are retained. SSU dataset, on the other hand, requires some means of sampling about equal amount of sequences from both sequence-rich and sequence-poor genera. Random sampling cannot do that because it doesn't take sequences or taxa into account, retaining (best case) and increasing (worst case) a clade size imbalance, rather than correcting it. Clearly, the requirements of SSU case study are extreme, but real analyses (like RuBisCo example in the Introduction) may be complicated by similar factors. Another weakness of RS is its stochastic nature, which makes the results of subsampling unpredictable. Other methods, being deterministic, are guaranteed to retain all the clades if sampling rate is sufficient. It may or may not be achievable for a given dataset. However, if a determenistic approach is able to retain 100% of Chs clades at 70% sampling rate, it will reliably retain 100% clades at any rate higher than 70%. Random sampling reaches only about 99% performance on the same data (Fig. 1b) because there are replicates lacking several clades by pure chance. Large and relatively distant clades of chitin synthase tree have guaranteed acceptable results of random sampling, but had an opposite effect on SS. The clades closest to query sequence are preferentially populated, while the rest of the tree remains undersampled even at the higher sampling rates (Fig. S1). This is not an issue for rRNA dataset: genera are small and close to each other, so SS retains about as many genera as DJ. AST performs well on the rRNA dataset, because this is exactly the case it was designed for: a large annotated dataset where clades in question are taxa. In fact, it performs perfectly under lower sampling rates: there are as many genera in reduced sets as there are sequences. Chitin synthase dataset is challenging for this method as taxonomy-based sampling is misleading when clades are not taxonomic, which is why AST stays behind both RS and DJ. Distant joining, on the other hand is performing perfectly on the chitin synthase dataset, sampling all clades even at the lowest sampling rate. On rRNA dataset it is second to AST, outperforming random sampling and SS.
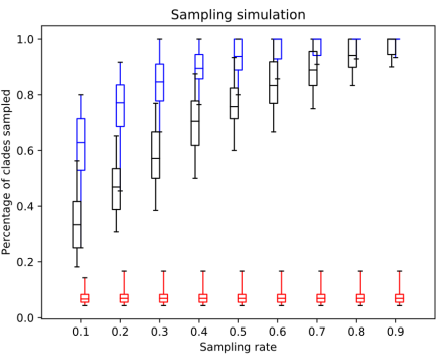
Fig. 2. The percentage of clades sampled by SS (red), random sampling (black) and distant joining (blue) in simulated trees.
6 Simulation results
The simulated datasets were designed to consist of a few oversampled clades and a number of small clades. This difference between clade sizes simulates the case when some part of the underlying tree is heavily sequenced and the rest is almost ignored. For example, these could be model groups like mammals or flowering plants, and a multitide of planctonic eukaryotic groups. Unsurprisingly, in these settings similarity sampling showed the worst performance. Since a random query sequence most likely comes from one of the larger clades, its closest neighbours are also from this clade, while the rest of the tree remains undersampled. Random sampling also misses a lot of clades for the same reasons it did with the Chs dataset. A clade's chance to be sampled is directly proportional to its size, and small clades in the presence of larger ones tend to be missed. Distance Joining, on the other hand, appears to be the best method, particularly with the lower sampling rates. Like AST in the rRNA dataset, this is precisely the case it was designed for: distant unequally-sized clades without available metadata. It does not achieve perfect sampling due to the sequence divergence within the large clades, but it outperforms all other approaches.
7 Conclusion
Similarity sampling performs worst on all data, whether real or simulated. It should be noted again that this approach is not discussed here as a reasonable sampling approach. Rather, it's an artifact that can arise from taking closest hits after search in some large databases. If the query sequnce belongs to a non-model taxon or an obscure subfamily of some complex protein family, the issue may be less pronounced. In general, though, applying an e-value or score threshold and taking all hits above it is a safer approach. If necessary, this collection of hits could be reduced to a practical size by one of the other methods. The choice of sampling method for a particular pipeline should depend on underlying assumptions that can be made about the data. If it is safe to assume there is no conflict between gene trees and the species tree and no unclassified (or incorrectly annotated) sequences are going to be included in the initial dataset, AST is the best option. The implementation by Zhou et al. can only work with the sequences that have Genbank IDs and depends on the NCBI taxonomy files, but the algorithm itself can be adapted for general case that do not violate aforementioned assumptions. If it's guaranteed that no clades of interest are represented by a handful of sequences, random sampling may be viable as well. It being random, there is always potential risk of missing/underrepresenting clades. However, with high enough sampling percentage and large enough clades this risk is minimal. Importantly, the exact sampling rate which is necessary for a given dataset, can be found by solving “generalized coupon collector's problem without replacements” (Wild et al. 2012). Additionally, random sampling has the advantage of being quick, requiring little additional memory, and not depending on the availability of external data. If none of the assumptions above can be made, distant joining is the safest approach. The only assumption it depends upon is purely integrated in the context of phylogenetic approach: the similarity of sequences tend to be higher within a clade, rather than between clades. Thus, assuming adequately generated distance matrix, DJ is designed to sample from different branches of the underlying tree independently of its topology and relative size of clades of interest, regardlessof any external data. There are, though, two limitations to this method: first, the results will be only as good as the distance matrix they were built on. Second, the computational cost of DJ spans the order of minutes for hundreds of sequences on a typical desktop. It is higher than for other methods discussed in this study. However, the time and computational requirements of the distance matrix construction, a most expensive step of DJ sampling, are still much less than those of the tree reconstruction. Thus, subsampling results in smaller data matrix for downstream analyses, which is beneficial for reduction of runtime at alignment and phylogenetic tree reconstruction steps . The performance of DJ can be further optimized by using fast distance estimation methods, such as alignment-free k-mer based approaches.
References
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. “Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB”. Appl Environ Microb, 72: 5069-5072
Huerta-Ceras J, Capella-Gutiérrez S, Pryszcz LP, Marcet-Houben M, Gabaldon T. 2014. “PhylomeDB v4: zooming into plurality of evolutionary histories of a genome”. Nucleic Acids Res. 42:D897-D902.
Morozov AA, Likhoshway YV. 2016. “Evolutionary history of the chitin synthases of eukaryotes”. Glycobiology doi: 10.1093/glycob/cww018.
Murray GGR, Weinert LA, Rhule EL, Welch JJ. 2016. “The phylogeny of Rickettsia using different evolutionary signatures: how tree-like is bacterial evolution?”. Syst Biol. 65(2): 265-279.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res. 41: D590-D596.
Rice P, Longden I, Bleasby A. 2000. “EMBOSS: the european molecular biology open software suite”. Trends in Genetics 16(6):276-277.
Saitou N, Nei M. 1987. “The neighbor-joining method: a new method for reconstructing phylogenetic trees”. Mol Biol Evol 4(4): 406-425.
Sonnhammer ELL, Hollich V. 2005. “Scoredist: a simple and robust protein sequence distance estimator”. BMC Bioinformatics 6:108.
Spielman SJ, Wilke CO. 2015. “Pyvolve: a flexible Python module for simulating sequences along phylogenies”. PloS ONE. 10(9): e0139047.
The UniProt Consortium. 2015. “Uniprot: a hub for protein information”. Nucl Acids Res 43: D204-D212.
Wild M, Janson S, Wagner S, Laurie D. 2012. “Coupon collecting and traversals of hypergraphs”. arXiv:1107.1401v3.
Zhou C, Mao F, Yin Y, Huang J, Gogarten JP, Xu Y. 2014. AST: and automated sequence-sampling method for improving the taxonomic diversity of gene phylogenetic trees. PLOS ONE 9(6): e98844.
References
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. “Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB”. Appl Environ Microb, 72: 5069-5072
Huerta-Ceras J, Capella-Gutiérrez S, Pryszcz LP, Marcet-Houben M, Gabaldon T. 2014. “PhylomeDB v4: zooming into plurality of evolutionary histories of a genome”. Nucleic Acids Res. 42:D897-D902.
Morozov AA, Likhoshway YV. 2016. “Evolutionary history of the chitin synthases of eukaryotes”. Glycobiology doi: 10.1093/glycob/cww018.
Murray GGR, Weinert LA, Rhule EL, Welch JJ. 2016. “The phylogeny of Rickettsia using different evolutionary signatures: how tree-like is bacterial evolution?”. Syst Biol. 65(2): 265-279.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res. 41: D590-D596.
Rice P, Longden I, Bleasby A. 2000. “EMBOSS: the european molecular biology open software suite”. Trends in Genetics 16(6):276-277.
Saitou N, Nei M. 1987. “The neighbor-joining method: a new method for reconstructing phylogenetic trees”. Mol Biol Evol 4(4): 406-425.
Sonnhammer ELL, Hollich V. 2005. “Scoredist: a simple and robust protein sequence distance estimator”. BMC Bioinformatics 6:108.
Spielman SJ, Wilke CO. 2015. “Pyvolve: a flexible Python module for simulating sequences along phylogenies”. PloS ONE. 10(9): e0139047.
The UniProt Consortium. 2015. “Uniprot: a hub for protein information”. Nucl Acids Res 43: D204-D212.
Wild M, Janson S, Wagner S, Laurie D. 2012. “Coupon collecting and traversals of hypergraphs”. arXiv:1107.1401v3.
Zhou C, Mao F, Yin Y, Huang J, Gogarten JP, Xu Y. 2014. AST: and automated sequence-sampling method for improving the taxonomic diversity of gene phylogenetic trees. PLOS ONE 9(6): e98844.