Bioinformatic analysis of conserved uncharacterized proteins in tailed bacteriophages
Bioinformatic analysis of conserved uncharacterized proteins in tailed bacteriophages
Abstract
The rapid development of genome sequencing technologies in recent decades has led to the accumulation of a huge amount of genetic information from a wide variety of organisms, from viruses to complex eukaryotes. This data flow has opened up unprecedented opportunities for understanding the fundamental principles of life organisation, evolutionary processes, and the molecular mechanisms underlying biological functions. However, despite significant progress in bioinformatic analysis and experimental research, a significant portion of the genes predicted from genomes still encode proteins whose function remains unknown or poorly characterised. Such proteins are commonly referred to as hypothetical proteins (HP) or uncharacterised proteins (UP)
, , .The aim of this work is to conduct a comprehensive review of the literature that highlights issues related to tools for annotating hypothetical proteins. The bacterium Klebsiella pneumoniae and the bacteriophages that infect it were used as model objects. This choice was justified in terms of both resistance and the relevance of bioinformatic tools. The article also discusses the work of key modern tools such as Pharokka, Panaroo, CD-HIT, HMMER, InterProScan, AlphaFold3, and Foldseek in relation to the selected model organisms, reflecting the features and shortcomings of these programmes.
The methodological part of the research is based on searching and analysing publications in leading databases such as Scopus, PubMed, Web of Science, Google Scholar, and SpringerLink, using keywords related to hypothetical proteins, annotation, homology comparison and search, pipeline, etc. Thus, this paper highlights the importance of finding tools suitable for annotating hypothetical proteins in widely distributed organisms that are important to humans.
1. Введение
Гипотетический белок — это белок, существование которого было предсказано, но для него отсутствуют экспериментальные доказательства того, что он экспрессируется в живых системах, то есть in vivo.
Масштаб проблемы гипотетических белков действительно велик. По оценкам, сделанным еще в середине 2000-х годов, в эпоху зарождения геномики, в большинстве полностью секвенированных геномов лишь для 50–70% генов удавалось предсказать функцию с высокой степенью достоверности . Эта методологическая преграда была настолько фундаментальной, что в научном сообществе стала известна как «70% барьер» . Эта проблема касалась не только экзотических или малоизученных организмов; даже для наиболее исследованных модельных систем, таких как Escherichia coli, значительная доля белков (до половины на тот момент) не имела экспериментально подтвержденной функции .
С тех пор количество секвенированных геномов выросло экспоненциально, но проблема «темной материи» генома, которую представляют гипотетические белки, не только сохранилась, но и усугубилась. Скорость выявления новых последовательностей с помощью технологий секвенирования нового поколения (NGS) значительно опережает скорость их функциональной характеризации , . Как следствие, доля неаннотированных генов в новых геномах остается стабильно высокой. Например, в недавних исследованиях геномов вирусов и бактериофагов отмечается, что до 65% вирусных генов остаются функционально неаннотированными , а в геномах фагов, инфицирующих Klebsiella pneumoniae, доля гипотетических или нехарактеризованных белков может составлять от 44% до более чем 60% от общего числа предсказанных генов , .
В свете этих трудностей особое внимание исследователей привлекают так называемые консервативные гипотетические белки (CHPs — Conservative Hypothetical Proteins) — белки, которые не имеют установленной функции, но обнаруживаются у филогенетически удаленных организмов или в пределах значительной группы родственных видов . Предполагается, что эволюционная консервативность таких белков свидетельствует об их функциональной значимости для клетки или вируса, так как бесполезные последовательности имели бы тенденцию быстро вырождаться под давлением мутаций. Именно поэтому CHPs рассматриваются как приоритетные цели для дальнейшего изучения, поскольку их характеризация может привести к открытию новых, ранее неизвестных биологических механизмов или функций, важных для понимания жизнедеятельности организмов .
Для приоритизации консервативных гипотетических белков (CHPs) с целью их дальнейшего экспериментального исследования был предложен ряд критериев. К ним относятся:
Широкое филогенетическое распространение: чем в большем количестве разнообразных геномов встречается CHP, тем выше вероятность его фундаментальной важности.
Эссенциальность (существенность): если данные нокаутных экспериментов (для клеточных организмов) показывают, что ген CHP является жизненно важным, это является сильным аргументом в пользу его значимости.
Данные экспрессии и взаимодействий: информация о том, что CHP экспрессируется при определенных условиях или взаимодействует с известными белками, может дать косвенные указания на его роль.
Наличие структурной информации: знание 3D-структуры белка, даже при отсутствии гомологии последовательностей, может позволить найти структурных аналогов с известной функцией.
Геномный контекст: анализ соседних генов (оперонная организация, консервативные генные кластеры) часто выявляет функциональные связи между белками.
Кроме того, была введена классификация CHPs на «известные неизвестные» (known unknowns) и «неизвестные неизвестные» (unknown unknowns). К первым относятся белки, для которых на основе слабых признаков гомологии, наличия определенных доменов или структурных особенностей можно предсказать общую биохимическую активность (например, АТФаза, протеаза, ДНК-связывающий белок), но их конкретная биологическая роль в клетке или вирусе остается неясной. «Неизвестные неизвестные» представляют собой белки, для которых отсутствуют какие-либо функциональные подсказки, что делает их наиболее сложными объектами для изучения, но потенциально и наиболее интересными с точки зрения открытия совершенно новых функций .
Именно поэтому задачей первостепенной важности в работе с такими белками становится функциональная аннотация — процесс присвоения им биологической роли. Этот подход основан на простом и логичном предположении: если два белка произошли от общего предка и сохранили схожую последовательность, то, скорее всего, они выполняют и схожую функцию. Тем не менее, этот метод имеет серьезные ограничения, которые и порождают обширный класс неаннотированных белков . Проблемы, связанные с аннотациями:
1. Если у гипотетического белка нет достаточно близких гомологов с уже известной функцией в базах данных, его аннотация становится затруднительной. Даже при наличии гомологии, функция не всегда сохраняется идентичной, особенно при значительной эволюционной дистанции, что может приводить к ошибочным предсказаниям.
2. Кроме того, многие гипотетические белки могут быть уникальными для определенных таксономических групп или даже для конкретных штаммов (так называемые ORFans – open reading frames), что делает поиск гомологов невозможным .
3. Другим существенным вызовом в аннотации нехарактеризованных белков является качество и полнота существующих баз данных . Ошибочные аннотации, однажды попавшие в базу, могут лавинообразно распространяться на другие последовательности через автоматизированные пайплайны переноса по гомологии.
4. Также базы данных могут быть неполными, особенно для менее изученных групп организмов или для белков с нетипичными функциями. Сложность усугубляется тем, что многие белки являются многофункциональными или участвуют в сложных, контекстно-зависимых взаимодействиях, что трудно отразить в простой функциональной метке .
Более того, экспериментальная проверка функций de novo для каждого гипотетического белка является чрезвычайно трудоемким, дорогостоящим и длительным процессом, что делает невозможным быструю характеризацию всех предсказанных ОРС .
2. Современные биоинформатические подходы к аннотации
Эти методы можно условно разделить на несколько категорий, часто используемых в комбинации для получения более надежных предсказаний , .
2.1. Методы, основанные на гомологии последовательностей
Наиболее фундаментальным и исторически первым подходом к функциональной аннотации является перенос функции по гомологии. Его центральная идея, заключается в том, что если белок с неизвестной функцией имеет значительное сходство последовательности с белком, функция которого уже установлена, то с высокой вероятностью они являются гомологами и выполняют схожую или идентичную функцию. Этот принцип лежит в основе большинства автоматизированных конвейеров аннотации.
Классическими инструментами для реализации этого подхода являются программы, выполняющие локальное выравнивание последовательностей.
BLAST (Basic Local Alignment Search Tool) и FASTA — это эвристические алгоритмы, которые стали основными методами молекулярной биологии. Они быстро сканируют гигантские базы данных (такие как GenBank или UniProt) в поисках последовательностей, имеющих статистически значимые совпадения с запросом. Их скорость достигается за счёт предварительного этапа, на котором они ищут короткие, идеально совпадающие «слова» (word matches), и только затем расширяют эти совпадения до полных локальных выравниваний. Несмотря на свою огромную пользу, BLAST и FASTA эффективны в основном для поиска относительно близких гомологов и могут упустить более отдаленные эволюционные связи, где сходство последовательностей со временем сильно снизилось , , .
Когда прямое сравнение последовательностей не даёт результатов, на помощь приходят более чувствительные методы, основанные на профилях, которые анализируют не одну последовательность, а целые семейства.
Профильные скрытые марковские модели (Profile HMMs) — это мощный статистический инструмент. HMM представляет собой не просто усреднённую последовательность, а сложную вероятностную модель, которая описывает характерные особенности целого семейства родственных белков. Она учитывает, какие аминокислоты являются критически важными и консервативными в определённых позициях, а какие — вариабельными, а также вероятности вставок и делеций. Это позволяет улавливать тонкие, но значимые паттерны, характерные для всего семейства, и находить очень отдалённых гомологов .
HMMER — это пакет программ, разработанный Шоном Эдди, который является золотым стандартом для работы с HMM-профилями. Он позволяет создавать HMM на основе выравнивания последовательностей и, что более важно, проводить сверхчувствительный поиск по базам данных профилей или сканировать последовательность на предмет совпадения с известными профилями . Именно на технологии HMM построены крупнейшие базы данных белковых доменов и семейств, которые являются ключевыми инструментами для глубокой функциональной аннотации.
Pfam — это, пожалуй, самая известная и широко используемая база данных белковых доменов. Она содержит обширную коллекцию тысяч курированных белковых семейств, каждое из которых представлено в виде множественного выравнивания и высококачественного HMM-профиля. Анализ белка с помощью сравнения с это базой данных позволяет определить его доменную архитектуру — то есть, из каких функциональных блоков он состоит. Поскольку домены часто являются носителями определённой биохимической активности, это даёт мощный ключ к пониманию общей функции белка . TIGRFAMs, SMART и PROSITE — это другие широко используемые базы данных, работающие по схожему принципу. TIGRFAMs фокусируется на белках прокариот и часто моделирует полноразмерные белки, а не только домены. SMART (Simple Modular Architecture Research Tool) специализируется на доменах, часто встречающихся в белках, участвующих в клеточном сигналинге. PROSITE уникальна тем, что помимо профилей она также содержит короткие, высококонсервативные паттерны (мотивы) и правила, которые могут с высокой точностью идентифицировать определённые функциональные сайты. Чтобы не сканировать каждую базу данных по отдельности, был создан интегрирующий ресурс: InterProScan — это программный пакет-агрегатор, который объединяет в себе поисковые возможности Pfam, TIGRFAMs, SMART, PROSITE и многих других баз данных. Запустив один поиск через InterProScan, исследователь получает исчерпывающий отчёт о всех известных доменах, семействах и функциональных сайтах, обнаруженных в его белке. Это делает InterProScan незаменимым инструментом для комплексной и глубокой функциональной аннотации гипотетических белков .
DIAMOND — это биоинформатический инструмент для быстрых и чувствительных поисков гомологии белковых последовательностей, аналогичный BLAST, но гораздо более производительный и оптимизированный для больших наборов данных. DIAMOND часто применяют для поиска близких и отдалённых гомологов в масштабных метагеномных и геномных данных. DIAMOND можно рассматривать как дополнительный модуль для быстрого предварительного поиска гомологов на начальных этапах (аналогично BLAST), особенно если работу нужно делать на больших объёмах данных, где BLAST может быть медленным. Таким образом, DIAMOND — популярный и мощный инструмент для поиска гомологии, он может выступать как вспомогательный или альтернативный инструмент, так и как основной метод .
2.2. Методы, основанные на анализе третичной структуры
Поскольку пространственная структура белка часто сохраняется в эволюции лучше, чем его первичная последовательность, и тесно связана с функцией, структурная биоинформатика играет все большую роль в аннотации гипотетических белков . До недавнего времени получение 3D-структуры было возможно в основном экспериментальными методами (рентгеноструктурный анализ, ЯМР), что ограничивало их применение. Однако революционный прорыв в предсказании структуры белков с помощью методов глубокого обучения, в первую очередь системы AlphaFold , кардинально изменил ситуацию. В настоящее время именно AlphaFold3 позволяет с высокой точностью предсказывать конформации для подавляющего большинства белков, включая гипотетические.
Полученная (экспериментально или предсказанная) 3D-структура затем может быть использована для поиска структурных аналогов в базе данных PDB (protein data base) с помощью таких инструментов, как Dali. Принцип его работы основан на сравнении матриц внутримолекулярных расстояний. Для каждого белка он строит матрицу, где записаны расстояния между всеми парами его Cα-атомов (атомов углерода, ближайших к функциональной группе). Затем он ищет в базе данных белки, у которых есть похожие по размеру и топологии блоки в этих матрицах. Этот подход очень надёжен и позволяет находить сложные, нетривиальные структурные сходства, однако он является достаточно медленным, что делает его менее удобным для крупномасштабных поисков или более современных и быстрых, как Foldseek. Его революционный подход заключается в преобразовании сложной третичной структуры в одномерную последовательность, используя так называемый структурный алфавит (3Di). Каждая «буква» этого алфавита описывает локальную геометрию основной цепи белка. Это позволяет свести задачу поиска по структурам к гораздо более быстрой задаче поиска по последовательностям. Благодаря этому Foldseek работает на порядки быстрее, чем Dali, сохраняя при этом сопоставимую чувствительность, что делает его идеальным инструментом для сканирования огромных баз данных предсказанных структур . Обнаружение структурного сходства с белком известной функции, даже при отсутствии значимой гомологии последовательностей свидетельствует о довольно высокой вероятности наличия аналогичной функции у этого белка.
2.3. Методы, основанные на белок-белковых взаимодействиях (PPI)
Белки редко функционируют в изоляции; в подавляющем большинстве случаев они являются частью сложных и динамичных сетей взаимодействий, выполняя свою роль в составе белковых комплексов или сигнальных каскадов. Данные о белок-белковых взаимодействиях (PPI) могут быть получены как экспериментально, с помощью высокопроизводительных методов (например, дрожжевая двугибридная система, ко-иммунопреципитация с последующей масс-спектрометрией для анализа белковых комплексов), так и предсказаны вычислительно на основе разнообразных данных.
Важным инструментом в этой группе методов является база данных STRING. Это не просто хранилище экспериментальных данных о PPI, а мощный интегративный ресурс, который агрегирует информацию из множества источников (каналов), включая экспериментальные данные, курируемые базы данных всевозможных (метаболических, биохимических и т.д.) путей, данные ко-экспрессии, текстовый майнинг научной литературы, а также информацию из методов геномного контекста, описанных выше. Каждому предсказанному взаимодействию в STRING присваивается балл уверенности, что позволяет оценить его надёжность . Анализ таких сетей взаимодействий с использованием алгоритмов кластеризации, таких как, например, Марковская кластеризация (MCL), позволяет выявлять плотно связанные группы белков, или функциональные модули. Если гипотетический белок попадает в один такой модуль вместе с белками известной функции, это является сильным аргументом в пользу его участия в том же биологическом процессе .
2.4. Методы, основанные на геномном контексте
Эти методы используют информацию о расположении генов в геноме и их совместном наследовании для предсказания функциональных связей. Принцип «вина по ассоциации» (guilt-by-association) предполагает, что гены, которые часто встречаются вместе в геномах (например, в одних и тех же оперонах или консервативных генных кластерах), вероятно, участвуют в одном и том же биологическом процессе или пути , . Для фагов, с их часто плотно упакованными и функционально организованными геномами, этот подход особенно актуален. Этот подход применяют для изучения белков фагов, так как их геном плотно упакован и функционально организован.
Анализ геномного контекста может включать:
- Соседство генов: идентификация генов, которые постоянно находятся рядом с геном интереса в разных геномах. Если соседи имеют известную функцию (например, гены лизиса, гены репликации), это может указывать на участие гипотетического белка в этом же процессе .
- Генные слияния (Rosetta Stone): это метод прогнозирования функции, основанный на событиях слияния белков. Два полипептида A и B в одном организме, вероятно, будут взаимодействовать, если их гомологи экспрессируются в виде одного полипептида AB в другом организме. Последний полипептид (AB) называется белком Розеттского камня, поскольку он содержит информацию как о A, так и о B. Этот метод может быть эффективным, поскольку биохимическая функция во многих случаях зависит от действия многомерного комплекса, демонстрирующего корреляцию между взаимодействующими белками и их функциями .
- Филогенетические профили: этот метод , анализирует паттерны присутствия/отсутствия генов (или их ортологов, то есть белков, произошедших от одной предковой формы) в наборе полностью секвенированных геномов. Гены со схожими филогенетическими профилями (т.е. они либо вместе присутствуют, либо вместе отсутствуют в одних и тех же геномах) с высокой вероятностью функционально связаны.
В то время как эти три метода (соседство, слияния и профили) предоставляют мощные, но разрозненные типы данных, современные подходы стремятся к их интеграции. Для этой цели разрабатываются специализированные инструменты, которые объединяют различные аспекты геномного контекста для более точной аннотации именно фаговых белков. Ярким примером такого инструмента является GOPhage (также упоминаемый в литературе как PhaGO). Этот конвейер не просто анализирует соседство генов, но и интегрирует эту информацию с данными, полученными из современных белковых языковых моделей. Он рассматривает геном как упорядоченное «предложение», где позиция гена и его «соседи» дают важные контекстуальные подсказки, что позволяет с высокой точностью предсказывать функциональные категории Gene Ontology даже для белков без явных гомологов .
2.5. Пан-геномный анализ
С появлением большого количества секвенированных геномов близкородственных штаммов или видов стал популярен пан-геномный анализ. Пан-геном включает в себя весь набор генов, встречающихся в данной таксономической группе, и делится на коровый геном (гены, присутствующие у всех или почти всех представителей), акцессорный геном (гены, присутствующие у некоторых) и уникальные гены. Этот подход позволяет перейти от анализа одного генома к изучению генетического разнообразия целой популяции.
Для построения пан-геномов используются специализированные инструменты, многие из которых основаны на графовых подходах, что позволяет более точно учитывать сложные структурные вариации в геномах , . Одним из таких современных инструментов является Panaroo. Его ключевое преимущество заключается в способности корректно работать с «шумными» данными, исправляя ошибки, возникающие из-за фрагментации сборок или контаминации. Panaroo строит граф, где узлами являются гены, а рёбрами — их соседство в геномах, что позволяет ему, например, «склеивать» гены, разорванные на два разных контига, и удалять гены-загрязнители (то есть случайно попавшие в образец и не являющиеся предметом исследования), не имеющие связей в графе. Для первичной и быстрой кластеризации схожих генов в ортологичные группы перед построением графа Panaroo использует такой эффективный инструмент, как CD-HIT . CD-HIT, в свою очередь, является широко используемой программой, которая реализует жадный алгоритм для быстрой кластеризации больших наборов последовательностей. Он итеративно группирует последовательности на основе заданного порога сходства, что позволяет значительно сократить избыточность данных и ускорить последующий анализ . Идентификация консервативных гипотетических белков (т.е. входящих в коровый геном, но не имеющих функции) является одной из важнейших задач пан-геномики, так как их повсеместное сохранение указывает на фундаментальную, но пока неизученную роль.
2.6. Методы машинного обучения (ML — Machine Learning) и глубокого обучения (DL — Deep Learning), включая белковые языковые модели (PLM — Protein Language Models)
В последние годы всё большее распространение получают методы, основанные на машинном и глубоком обучении, для предсказания функций белков. Эти подходы способны извлекать сложные, нелинейные закономерности из больших наборов данных (последовательности, структуры, домены, взаимодействия и т.д.) и строить на их основе высокоточные предсказательные модели. Разрабатываются инструменты, которые стремятся к интеграции разнородной информации. Примером такого подхода является DPFunc. Этот инструмент использует архитектуру глубокого обучения для объединения двух ключевых типов информации: информации о доменах, которая даёт общее представление о функциональных блоках белка, и информации о его третичной структуре (часто предсказанной с помощью AlphaFold3), которая предоставляет детальные пространственные данные. Интегрируя эти два типа информации о белках (доменная и третичная структуры), DPFunc достигает высокой точности предсказаний и, что важно, обеспечивает интерпретируемость, подсвечивая ключевые домены и остатки, вносящие наибольший вклад в предсказанную функцию .
Особый интерес в рамках этого направления представляют белковые языковые модели (PLM — Protein Language Models). Эти модели, такие как ESM или ProtBERT, обучаются на огромных массивах данных белковых последовательностей (сотни миллионов или даже миллиарды) по принципу, схожему с большими языковыми моделями типа GPT. Они учатся предсказывать «скрытые» аминокислоты в последовательности, что позволяет им улавливать глубинные эволюционные, структурные и функциональные закономерности «языка» белков. В результате PLM генерируют для каждой последовательности богатое информационное векторное представление (embedding). Эти «эмбеддинги» затем могут использоваться как входные данные для более простых моделей машинного обучения для решения различных задач, включая предсказание функций. PLM показывают огромный потенциал, особенно для аннотации белков без явных гомологов, так как они улавливают контекст, а не только прямое сходство .
Таким образом, современная стратегия аннотации гипотетических белков всё чаще опирается на интеграцию информации из различных источников и применение комбинации вычислительных подходов. Это позволяет постепенно сокращать долю «темной материи» в геномах и получать более полное представление о биологических системах.
3. Выбор модельной системы
Выбор модельной системы для изучения гипотетических белков является ключевым этапом любого исследования. В идеале такой организм должен соответствовать ряду требований: иметь полностью секвенированный и хорошо аннотированный геном, быть доступным для генетических манипуляций и представлять интерес для фундаментальной или прикладной науки. Однако в свете глобального кризиса антибиотикорезистентности фокус исследователей всё чаще смещается на клинически значимые патогены, изучение которых может дать немедленный практический результат.
Одним из таких приоритетных объектов, отвечающих требованиям актуальности и важности, является Klebsiella pneumoniae. Эта бактерия является не просто удобной моделью, а представляет собой одну из самых серьезных угроз для глобального здравоохранения. Она входит в группу ESKAPE-патогенов, печально известных своей способностью вызывать тяжелые внутрибольничные инфекции и формировать множественную лекарственную устойчивость. Распространение штаммов, резистентных к антибиотикам «последнего резерва», делает поиск альтернативных методов борьбы с этим патогеном задачей первостепенной важности. В этом контексте изучение её бактериофагов и их геномного «арсенала», включая гипотетические белки, приобретает особое значение. Понимание того, как функционируют эти вирусы и их белки, может стать ключом к разработке новых терапевтических стратегий. Таким образом, выбор Klebsiella pneumoniae и её фагов в качестве системы для исследования обусловлен их неоспоримой клинической значимостью и острой необходимостью поиска новых путей борьбы с этим опасным патогеном .
3.1. Klebsiella pneumoniae — условно патогенная бактерия
Klebsiella pneumoniae представляет собой грамотрицательную палочковидную бактерию из семейства Enterobacteriaceae, которая широко распространена в окружающей среде, а также является комменсалом слизистых оболочек желудочно-кишечного тракта и верхних дыхательных путей человека и животных. Однако, при определенных условиях, особенно у лиц с ослабленным иммунитетом или при нарушении целостности естественных барьеров, K. pneumoniae способна вызывать широкий спектр тяжелых инфекционных заболеваний, что делает ее одним из наиболее значимых оппортунистических патогенов . Спектр вызываемых инфекций включает пневмонию (особенно вентилятор-ассоциированную), инфекции мочевыводящих путей, раневые инфекции, сепсис, менингит и абсцессы печени . K. pneumoniae входит в группу так называемых ESKAPE-патогенов (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter species), которые представляют наибольшую угрозу в связи с их высокой вирулентностью и способностью быстро приобретать устойчивость к антимикробным препаратам .
Патогенный потенциал K. pneumoniae обусловлен наличием у нее ряда факторов вирулентности. Ключевую роль играет капсульный полисахарид (CPS), который покрывает бактериальную клетку, защищая ее от фагоцитоза макрофагами, действия системы комплемента и некоторых антибиотиков, а также способствуя адгезии к тканям хозяина . Существует более 77 серологически различных капсульных типов K. pneumoniae, и некоторые из них (например, K1, K2) ассоциированы с гипервирулентными штаммами, способными вызывать инвазивные инфекции даже у иммунокомпетентных лиц. Другими важными факторами вирулентности являются липополисахарид (LPS), являющийся эндотоксином, сидерофоры (например, энтеробактин, сальмохелин, аэробактин, иерсиниабактин), которые обеспечивают бактерии железом в условиях его дефицита в организме хозяина, и фимбрии (пили), участвующие в адгезии к эпителиальным клеткам , .
Особую проблему представляет способность K. pneumoniae формировать биопленки на различных абиотических (медицинские катетеры, импланты) и биологических поверхностях , . Биопленки — это структурированные микробные сообщества, заключенные в самопродуцируемый матрикс из экзополисахаридов, белков и внеклеточной ДНК. Находясь в биопленке, бактерии значительно более устойчивы к действию антибиотиков и факторов иммунной системы хозяина, что делает инфекции, связанные с биопленками, особенно трудноизлечимыми . Формирование биопленок также способствует персистенции патогена и может служить резервуаром для горизонтального переноса генов, включая гены антибиотикорезистентности .
3.2. Проблема антибиотикорезистентности Klebsiella pneumoniae
Одной из наиболее серьезных угроз, связанных с K. pneumoniae, является ее стремительно растущая и распространяющаяся устойчивость к антимикробным препаратам. Эта бактерия обладает способностью быстро приобретать гены резистентности к широкому спектру антибиотиков, включая бета-лактамы (в том числе карбапенемы — антибиотики «последнего резерва»), аминогликозиды, фторхинолоны и полимиксины , . Распространение штаммов, продуцирующих бета-лактамазы расширенного спектра (БЛРС, ESBL), которые гидролизуют большинство пенициллинов и цефалоспоринов, стало глобальной проблемой. Еще большую озабоченность вызывает появление и распространение штаммов K. pneumoniae, резистентных к карбапенемам (Carbapenem-Resistant Klebsiella pneumoniae, CRKP), основной механизм устойчивости которых связан с продукцией карбапенемаз (например, KPC, NDM, OXA-48-подобные) , . Инфекции, вызванные CRKP, характеризуются высокой летальностью из-за крайне ограниченных терапевтических возможностей. Механизмы приобретения резистентности у K. pneumoniae разнообразны и включают как мутации в собственных генах, так и, что наиболее важно, горизонтальный перенос генов (ГПГ) резистентности, часто локализованных на мобильных генетических элементах, таких как профаги, плазмиды и транспозоны . Это обеспечивает быстрое распространение множественной лекарственной устойчивости не только между штаммами K. pneumoniae, но и к другим видам бактерий.
3.3. Бактериофаги как потенциальная альтернатива в борьбе с Klebsiella pneumoniae
Нарастающая проблема антибиотикорезистентности, особенно у таких патогенов, как K. pneumoniae, остро ставит вопрос о необходимости поиска и разработки альтернативных антибактериальных стратегий . Одним из наиболее перспективных направлений является фаготерапия — использование бактериофагов (фагов), вирусов, специфически инфицирующих и лизирующих бактериальные клетки, для лечения бактериальных инфекций. Жизненные циклы бактериофагов в основном схожи, но у некоторых он протекает без перерывов (литический цикл), а у некоторых фаговая ДНК встраивается в геном бактерии и никак себя не проявляет в течение многих поколений (профаг). Но в определенный момент времени ДНК профага высвобождается, завершает свой жизненный цикл, что приводит к гибели клетки хозяина (литический цикл).
Бактерии используют различные антизащитные системы для защиты от бактериофагов, смысл которых уничтожить ДНК фага любым способом. Некоторые из них: система рестрикции-модификации (R-M), система CRISPR-Cas, и так далее. Одной из самых известных защитных систем является CRISPR-Cas, в которой бактерия узнает специфический участки фаговой ДНК с помощью гидовой РНК и вырезает эту чужеродную для нее ДНК, сшивая образовавшиеся бреши ДНК друг с другом.
Фаги обладают рядом преимуществ перед антибиотиками:
- Высокая специфичность: Фаги обычно инфицируют только определенные виды или даже штаммы бактерий, не затрагивая нормальную микрофлору хозяина, что снижает риск дисбиозов , .
- Способность к саморепликации: Фаги могут размножаться в месте инфекции до тех пор, пока присутствуют чувствительные бактерии-хозяева.
- Активность против биопленок: Некоторые фаги или их ферменты (например, деполимеразы) способны разрушать матрикс биопленок, делая бактерии внутри них доступными для лизиса или действия других антимикробных агентов
, .- Разнообразие и доступность: Фаги являются самыми многочисленными биологическими объектами на планете и могут быть выделены из различных природных источников.
Исследования по выделению и характеризации фагов, активных против K. pneumoniae, ведутся во многих лабораториях мира, что отражено в растущем числе публикаций . Были описаны фаги K. pneumoniae, принадлежащие к различным семействам хвостатых фагов (например, Myoviridae, Siphoviridae, Podoviridae, а также их современные таксономические эквиваленты в рамках класса Caudoviricetes), демонстрирующие литическую активность против клинических изолятов, включая мультирезистентные штаммы и штаммы, образующие биопленки , , , . Для повышения эффективности и расширения спектра действия, а также для снижения вероятности развития бактериальной резистентности к фагам, часто предлагается использование фаговых коктейлей, состоящих из нескольких различных фагов .
3.4. Важность геномного анализа бактериофагов для терапевтического применения
Несмотря на большой потенциал, использование фагов в терапии требует их тщательной характеризации, ключевым элементом которой является полный геномный анализ , , . Секвенирование и аннотация фагового генома позволяют:
- Определить жизненный цикл фага: литические фаги, вызывающие гибель бактериальной клетки, являются предпочтительными для терапии. Необходимо исключить умеренные фаги, способные к лизогении, так как они могут интегрировать свою ДНК в геном бактерии и переносить гены, в том числе гены вирулентности или резистентности , .
- Оценить безопасность: геномный анализ позволяет выявить наличие генов, кодирующих известные токсины, факторы вирулентности или гены антибиотикорезистентности, которые могли бы быть переданы бактерии-хозяину, что недопустимо для терапевтических фагов , , .
- Идентифицировать полезные гены: в геномах фагов могут быть обнаружены гены, кодирующие ферменты с антибактериальной активностью, такие как эндолизины (разрушают пептидогликан) или капсульные деполимеразы (разрушают капсулу бактерии, делая ее более уязвимой), которые сами по себе могут рассматриваться как терапевтические агенты (энзибиотики) , .
- Понять механизмы взаимодействия с хозяином: аннотация генов, отвечающих за адсорбцию, репликацию, сборку вирионов, помогает понять биологию фага.
Однако, как уже отмечалось в предыдущем разделе и подтверждается многочисленными исследованиями фаговых геномов , , , , значительная часть предсказанных генов в геномах фагов кодирует гипотетические белки с неизвестными функциями. Эта «темная материя» фаговых геномов может скрывать как новые полезные функции, так и потенциально нежелательные, что подчеркивает актуальность их детального биоинформатического анализа и функциональной аннотации.
4. Концепция интегративного пайплайна для анализа гипотетических белков
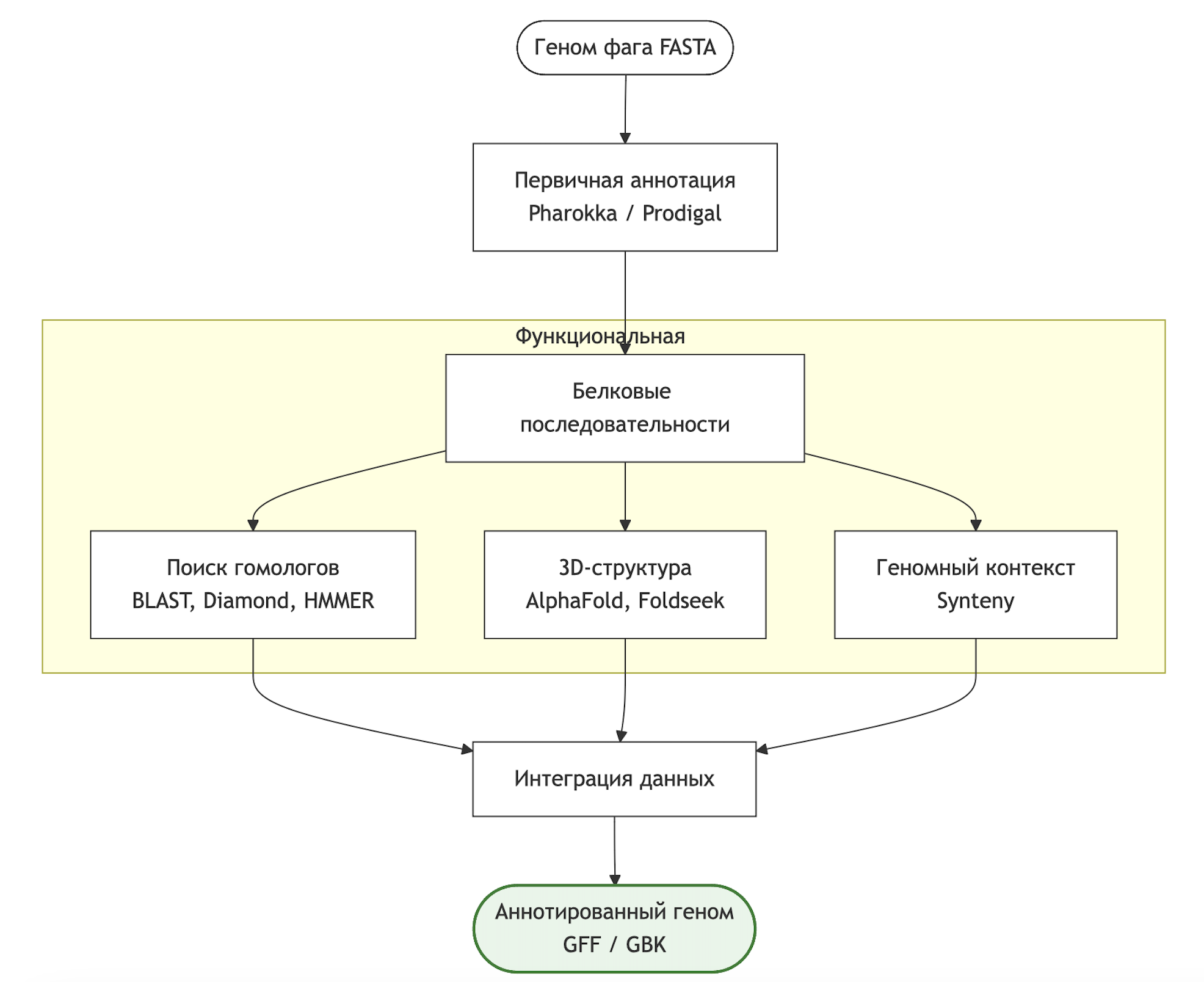
Рисунок 1 - Схема предлагаемого интегративного подхода к аннотации гипотетических белков
- Пан-геномный анализ: сравнение геномов группы фагов K. pneumoniae с помощью инструментов, основанных на графовых подходах, таких как Panaroo , позволяет идентифицировать консервативные гипотетические ортогруппы (КГО). Такие КГО, присутствующие у многих фагов данной группы, являются особенно интересными кандидатами для дальнейшего изучения, так как их консервативность предполагает важную, но пока неизвестную функцию. Для предварительной кластеризации белков в таких пайплайнах часто используется CD-HIT . Теоретические основы и преимущества графовой пан-геномики подробно рассмотрены в обзорах , .
- Глубокий поиск гомологии и доменов: для белков из КГО необходимо провести углубленный поиск гомологов с использованием чувствительных методов, таких как HMMER , по базам данных профилей (например, Pfam ) и комплексный анализ доменной архитектуры с помощью InterProScan . Известно, что геном бактериофагов обладает модульной организацией. То есть геном фагов «мозаичен» — сшит из многочисленных модулей, каждый из которых обладает собственной эволюционной историей. И потому такая модульная природа многих фаговых белков делает доменный анализ особенно важным .
- Предсказание пространственной структуры: программа AlphaFold3 , открыла возможность получать высокоточные 3D-модели для подавляющего большинства белков, включая гипотетические. Для фаговых белков это особенно актуально, так как экспериментального подтверждения для них относительно мало. Полученные модели затем можно сравнивать с известными структурами из PDB с помощью серверов Dali или Foldseek для поиска структурных аналогов, что может дать ключ к пониманию функции.
- Анализ геномного контекста: изучение генов, соседствующих с генами, кодирующими гипотетические белки, в геномах фагов K. pneumoniae, может выявить их функциональную связь с известными генами (например, участие в лизисе, репликации, сборке). Специализированные инструменты, такие как GOPhage/PhaGO (см.выше) разрабатываются для автоматизации такого анализа.
Интеграция данных и формулировка гипотез: конечной целью биоинформатического анализа является интеграция всех полученных данных (гомология, домены, структура, контекст, филогенетические связи) для формулировки обоснованных гипотез о возможных функциях исследуемых гипотетических белков.
5. Заключение
Применение такого комплексного подхода на практике сталкивается с серьезным методологическим вызовом. Каждый из этапов анализа, такие как, пангеномика, сборка, аннотация и структурное моделирование, требует использования специализированных, часто не связанных друг с другом программных инструментов. Исследователю приходится вручную запускать каждый инструмент, форматировать выходные данные и передавать их на вход следующей программе, что не только отнимает много времени, но и является источником потенциальных ошибок. На сегодняшний день не существует единого комплексного решения, которое бы интегрировало эти передовые методы в единый, автоматизированный рабочий процесс (пайплайн) для аннотации гипотетических белков фагов. Кроме того, в литературе отсутствуют данные о наличии нового программного конвейера, реализуемого в виде скрипта, который бы автоматизировал весь процесс глубокой аннотации гипотетических белков у фагов, инфицирующих Klebsiella pneumoniae. Согласно обзору литературы, не существует какого-либо программного конвейера, который бы последовательно вызывал и управлял работой ключевых современных инструментов (Pharokka, Panaroo, CD-HIT, HMMER, InterProScan, AlphaFold3, Foldseek и др.), а также агрегировал их результаты и представлял их в виде сводного отчета. Создание такого инструмента позволит перейти от фрагментарного ручного анализа к целостному, высокопроизводительному и воспроизводимому подходу, что кардинально ускорит процесс исследования «темной материи» фаговых геномов.