МОЛЕКУЛЯРНАЯ ТАКСОНОМИЯ И ЭВОЛЮЦИОННОЕ УЧЕНИЕ В СВЕТЕ НОВЕЙШИХ БИОИНФОРМАТИЧЕСКИХ И КОСМОЛОГИЧЕСКИХ ДАННЫХ

MOLECULAR TAXONOMY AND THE EVOLUTION THEORY IN LIGHT OF EMERGING BIOINFORMATIC AND COSMOLOGICAL DATA
Acknowledgements
Thanks to Mr. Dmitry N. Tychinin (IBPPM RAS) for translating the original manuscript into English.
Conflict of Interest
None declared.
Shchyogolev S.Yu.
* Institute of Biochemistry and Physiology of Plants and Microorganisms, Russian Academy of Sciences, 13 Entuziastov Avenue, Saratov, 410049, Russia; Chernyshevsky Saratov State University, 83 Astrakhanskaya Street, Saratov, 410012, Russia
*To whom correspondence should be addressed.
Associate Editor: Giancarlo Castellano Received on 07 July 2016;
revised on 01 September 2016; accepted on 15 September 2016
Abstract
A brief review is given of recent advances in the taxonomic study of organisms and current views on biological evolution and the origin of life. A discussion is presented on how the treelike and net components contribute to the topology of phylogenetic constructs, with account taken of the prevailing role of horizontal gene transfer in prokaryote evolution and life. Approaches are described to the rational selection and practical use of phylogenetic markers (including 16S rRNA gene DNA sequences) in biomedical (including metagenomic) applications with traditional and nontraditional (large) amounts of molecular genetic data. Emerging results from taxonomic studies of the Earth’s biota and the methods of their generation are demonstrated. It is noted that the current developments in particle physics and in cosmology have important implications for solving paradoxes associated with the vanishingly small probability of some fundamental processes of prebiological and biological evolution.
Keywords: taxonomy, phylogenetic tree, horizontal gene transfer, 16S rRNA, big data, metagenomics, nonculturable prokaryotes, biological and prebiological evolution, eternal chaotic inflation, multiverse.
Contact: shegolev_s@ibppm.ru
1. Introduction
A principal sphere in which bioinformation resources are applied is taxonomic research, which uses the phylogenetic characteristics of the Earth’s organisms to establish kinship and evolutionary interrelations among them. Phylogenetic taxonomy, including molecular phylogenetics, is an effective tool in biodiversity studies (Hug et al., 2016), ecoresearch, identification of human-beneficial or harmful organisms (Costello et al., 2013), and other biomedical and biotechnological applications. This research creates a basis for significant progress in views on the evolution and origin of living nature, and this progress is ensured, among other factors, by advances in comparative genomics (Koonin, 2012) with the use of emerging particle physics and cosmological data (Linde, 2014; Susskind, 2006; Vilenkin, 2006).
From senior (domains) to junior (species) ranks, the number of taxonomic units within individual ranks increases. According to a recent study (Costello et al., 2013), which appears to be sufficiently accurate, the number of species living on Earth is 5±3 million, of which 1.5 million species are named. The current estimates of the number of predicted species also vary broadly (Mora et al., 2011; Table 1). As of 2014, the existing nucleotide sequence databases contain 6×1011 bases (Lesk, 2014), corresponding to 200 complete human genomes ( 3×109 bases per genome). The total amount of base pairs on Earth (global biodiversity) is 5×1037 (https://en.wikipedia.org/wiki/Global_biodiversity). In other words, the upper limit of bioinformation resources for this (major) type only is equivalent to the sum of the complete genomes of approximately 1028 Homo sapiens individuals. Developing effective means to organize, maintain, improve, and navigate these resources, as well as to extract useful data from them, which are in demand in biomedicine and related areas, necessitates accounting for the specific character of manipulations with large data arrays. These activities are termed the “big data problem” (http://bigdatawg.nist.gov/home.php), and the subject of this review-topical taxonomic and evolutionary research using a large arsenal of available molecular genetic data-readily illustrates it.
3×109 bases per genome). The total amount of base pairs on Earth (global biodiversity) is 5×1037 (https://en.wikipedia.org/wiki/Global_biodiversity). In other words, the upper limit of bioinformation resources for this (major) type only is equivalent to the sum of the complete genomes of approximately 1028 Homo sapiens individuals. Developing effective means to organize, maintain, improve, and navigate these resources, as well as to extract useful data from them, which are in demand in biomedicine and related areas, necessitates accounting for the specific character of manipulations with large data arrays. These activities are termed the “big data problem” (http://bigdatawg.nist.gov/home.php), and the subject of this review-topical taxonomic and evolutionary research using a large arsenal of available molecular genetic data-readily illustrates it.
Table 1. Numbers of catalogued and predicted species on Earth and in the Ocean
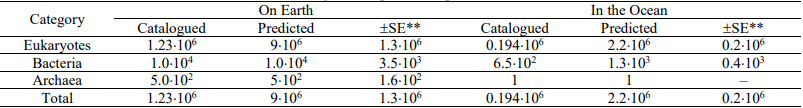
* Adapted from Mora et al. (2011)
** Standard error for the number of predicted species.
(Figure 1) shows a canonical phylogenetic tree that reflects taxonomic interrelations at the level of senior ranks: domains 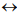 kingdoms
kingdoms 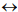 phyla
phyla 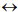 classes
classes 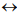 orders. As a phylogenetic marker, the treelike portion of this scheme uses the molecular sequences of the DNA of the 16S/18S rRNA gene, belonging to “informational” genes (Koonin, 2012), which control replication of DNA, transcription, and translation. The arrows mark additional linkages between taxonomic groups, introduced by possible exchange of genetic material between different species (horizontal gene transfer, HGT). These include species that do not form part of the same treelike fragments of the phylogenetic constructs combining groups of organisms originating from common ancestors and determined by vertical gene transfer (from ancestors to descendants). HGT is most common in prokaryotes, in most of which it is responsible for the formation of the overwhelming majority of genomes during evolution (Koonin, 2012). HGT continues to contribute substantially to the acquisition of diverse traits by prokaryotes as they adapt to their ecological niches (Coenye et al., 2005) (and microbial diversity on the whole).
orders. As a phylogenetic marker, the treelike portion of this scheme uses the molecular sequences of the DNA of the 16S/18S rRNA gene, belonging to “informational” genes (Koonin, 2012), which control replication of DNA, transcription, and translation. The arrows mark additional linkages between taxonomic groups, introduced by possible exchange of genetic material between different species (horizontal gene transfer, HGT). These include species that do not form part of the same treelike fragments of the phylogenetic constructs combining groups of organisms originating from common ancestors and determined by vertical gene transfer (from ancestors to descendants). HGT is most common in prokaryotes, in most of which it is responsible for the formation of the overwhelming majority of genomes during evolution (Koonin, 2012). HGT continues to contribute substantially to the acquisition of diverse traits by prokaryotes as they adapt to their ecological niches (Coenye et al., 2005) (and microbial diversity on the whole).
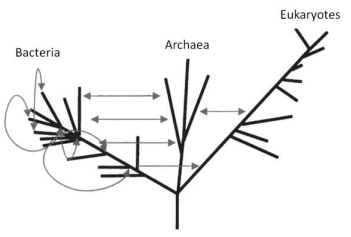
Fig. 1 - Canonical view of the phylogenetic tree of life (dark lines) combined with netlike topology of phylogenetic constructs (arrows), reflecting horizontal gene transfer.
Adapted from Woese et al. (1990) and Fraser-Liggett (2005)
The topology of the treelike portion of the generalized phylogenetic tree (Koonin, 2012), determined by the basic mechanisms of DNA replication and cell division, is tied to mutations occurring mostly in highly conserved informational genes. Conversely, its netlike structure, determined by the ubiquitous occurrence of HGT in prokaryotes, is linked to transformations occurring mostly in operational genes, which control cell metabolism, synthesis of membrane proteins and signal molecules, and other processes (Koonin, 2012).
The demonstration that HGT plays a dominant role in the evolutionary development and life of prokaryotes is a principal conceptual achievement in comparative genomics (Koonin, 2012). It necessitates a fresh look at the significance of the taxonomic constructions obtained by traditional molecular genetic methods. Specifically, this demonstration leads to the estimation of the possibility in principle and the range of applicability to these constructions of the treelike (vertical) topology of phylogenetic plots of the type in (Figure 1). On the basis of multifaceted, thorough investigations summarized by Koonin (2012), in the relatively small group of about a hundred informational genes (among the approximate total number of tens of thousands of genes in specific organisms), a set of nearly universal trees (NUTs) have been revealed that have a topology reflecting mostly a treelike evolutionary history. Of basic importance is the ascertaining of the fact that the consensual topology of NUTs almost repeats the 16S rRNA tree. This gives additional grounds to consider the 16S rRNA tree to be a major marker of “vertical” phylogeny (Woese, 2002).
The significance of HGT weakens considerably in the evolutionary development of eukaryotes, although early in biological evolution, the most probable mechanism of the rise of eukaryotes is active HGT (entrapment in the bacterial and archaeal suprakingdoms of some organisms of different species by other organisms and their subsequent symbiotic coexistence with appropriate redistribution of genetic material). Quite possibly, organelles such as mitochondria, chloroplasts, and cell nuclei were acquired by eukaryotes via the symbiotic mechanism (Koonin, 2012).
2. 16S rRNA technology in studies using traditional and nontraditional (large) amounts of molecular genetic data
Because the corresponding genes are conserved sufficiently highly, 16S/18S rRNA technology not only serves as a source of valuable information about the origin and evolutionary history of species but also allows prediction of the physiological–biochemical properties of the organisms being discovered de novo. As a guide, one can use the properties of interest possessed by the organisms that are part of the taxonomic environment of the newly discovered species. This is a fairly established practice in biomedicine and biotechnology.
The development and wide application of 16S rRNA technology has significantly promoted a sharp increase in studies on the discovery and identification of new prokaryotic species (Oren, Garrity, 2014) and an expansion of biomedical (Chakravorty et al., 2007) and metagenomic (Biteen et al., 2016) research. These studies deal with the isolation and separation of the total genetic material from natural objects, which contains tens and hundreds of thousands of genetic sequences to analyze. Another promoting factor was that next-generation equipment for DNA sequencing and amplification had entered the market (Chun, Rainey, 2014).
The steady increase in deciphered genetic structures, including those used in taxonomy, generates conditions (and need) for substantial increase in taxonomic units of different ranks (from species, genera, and families to orders, classes, and phyla) that are simultaneously included in phylogenetic constructs. However, these constructs cannot be effectively visualized within classic phylogenetic schemes of the type in (Figure 1) (visible enough, with the number of branches and nodes not greater than on the order of several tens) when the number of taxonomic units being considered is large enough (on the order of thousands or even greater than that). This is attested by the phylogenetic diagram at http://www.zo.utexas.edu/faculty/antisense/download-filestol.html , which attempts to illustrate the evolutionary interrelations among approximately 3,000 species (0.2% of the approximately 1.5 million known species). Thus, there is a substantially increasing need to apply and further develop special means of statistical analysis to ensure that the big taxonomic data being processed are visualized in a more acceptable and researcher-friendly form.
Chun and Rainey (2014) illustrate how this problem can be solved for 10,944 prokaryotic species ( 60 million pairwise comparisons of 16S rRNA genes) on the basis of principal coordinate analysis. The clustering of bacterial species by this method and their diversity (see http://www.ezbiocloud.net/ezgenome/status) reveal clearcut phylogenetic distinctions between firmicutes and actinobacteriatwo phyla previously identified as one taxonomic group of gram-positive bacteria in the classic tree of life (Woese et al., 1990). A similar approach was adopted for the clustering of phylogenetic constructs and detection of NUT groups within the Forest of Life concept described by Koonin (2012).
Ruan et al. (2012; see also http://salsahpc.indiana.edu/millionseq/mina/16SrRNA_index.html) describe and cite literature on an alternative data clustering approach, DACIDR (deterministic annealed clustering with interpolative dimension reduction). This approach considers the sequencing results for about a million 16S rRNA gene sequences. It ensures that the original high-dimensional data are transformed into target dimension space (2D, 3D), with the picture of distribution of evolutionary distances in the original space being preserved with the greatest accuracy possible. The method requires the use of high-performance commercial computing systems with about 800 processor cores. For a comparative analysis of several methods for species clustering according to genetic sequences, including identification of operational taxonomic units, see Chen et al. (2013) and references at http://omictools.com/binning-16sdatasets-category.
As an illustration, the website http://salsahpc.indiana.edu/millionseq/mina/16SrRNA_index.html shows the results of clustering and visualization of large sets of random 16S rRNA data from the Human Microbiome Project (The Human Microbiome Project Consortium, 2012). Also shown are the results of application of this technology to phylogenetic metagenomic investigations of fungi by using gene sequences of ribosomal 28S rRNA.
Thus, the approaches being developed to the 2D and 3D visualization of taxonomic interrelations by using large arrays of molecular genetic data (Figure 2) are of basic and practical interest. Rapid and reliable transformation of such data into friendly visualizations can ensure recognition of previously unnoticed interrelations (or differences, see above) among taxonomic groups and a more reliable prediction of the physiological–biochemical properties of organisms, a deeper understanding of evolution, and estimation of the effectivity of biotechnological and biomedical means and resources. This is particularly attractive in view of the methods being actively developed for the computer implementation of these tasks in the interactive and local modes.
3. Alternative phylogenetic markers and their use in emerging taxonomic studies of the Earth’s biota
The steady increase in the sizes of bioinformation resources creates prerequisites for genotypic approaches in systematics that use information about sequences from the ever-widening parts of organismal genomes (ideally whole genomes), which, in principle, brings bioinformatic estimates closer to the experimental results from total DNA– DNA hybridization tests (Oren, Garrity, 2014). However, whole-genome sequencing on a mass scale remains fairly complex and costly even in prokaryotes, for which metagenomic studies using a large number of genomic fragments for various nonculturable microflora members are increasingly becoming of high priority (Hug et al., 2016). In this case, multilocus sequence analysis (MLSA) based on fairly conserved “housekeeping” genes (De Vos, 2011) is appropriate to use.

Fig. 2 - 3D visualization (projection onto the plane) of the diversity and clustering of culturable (1) and nonculturable (2) bacterial species as a result of 16S rRNA gene sequencing.
Adapted from http://www.ezbiocloud.net/ezgenome/status
Another promising alternative to 16S/18S rRNA technology is the determination of the average nucleotide identity (ANI; Kim et al., 2014). The ANI deals with homologous areas shared by two genomes and is the possible “gold standard” instead of experimental DNA–DNA hybridization (Oren, Garrity, 2014). The need for such a replacement has been pointed out by many researchers in view of the noticeably decreased popularity of DNA–DNA hybridization owing to its labor-intensive nature and relatively low accuracy. Kim et al. (2014) compared ANI and 16S rRNA gene sequence similarity by using ~ 7,000 prokaryotic genomes from 22 phyla (more than a million pairwise comparisons), and they proposed a corrected boundary value for species delineation in a 16S rRNA test: 98.65% sequence identity versus the traditionally accepted 97% (Tindall et al., 2010). High correlation was observed between the ANI value and the DNA–DNA hybridization percentages, and it was found that the commonly accepted cutoff value of 70% in a DNA–DNA test for the species identification of an isolate, as compared with the known reference species, corresponds to 95–96% ANI (Kim et al., 2014).
An example of an MLSA-based approach comes from one of the most impressive publications of recent time, which appears to propose a new view of the tree of life and the diversity of the Earth’s biota (Hug et al., 2016). The major methodological novelty in that work was the extended (as compared with 16S/18S rRNA technology) use of markers coded for by informational genes-amino acid sequences of ribosomal proteins concatenated into sets of 16 specially selected proteins. Account was taken of the data only for those organisms whose genomes were estimated to be (almost) complete on the basis of their sets of genetic sequences. This increased the reliability of genomic and metagenomic results, improved the resolution capacity of the phylogenetic constructs, and provided additional information about the presence or absence of specific metabolites in the organisms. Clearly, this approach agrees with the principles of evaluation of the phylogenetic markers that most correctly reflect the “treelike” evolutionary history of organisms (Koonin, 2012).
Hug et al. (2016) used genomic data on more than 1,000 nonculturable and little known organisms and on more than 3,000 genomes from publicly available bioinformatic databases. In the former case, the data stemmed from a wide range of environmental samples: from aqueous, terrestrial, and subterrestrial ecosystems to animals. The resultant tree of life included 92 named bacterial phyla, 26 archaeal phyla, and all the 5 eukaryotic supergroups known to date. Generation of complete phylogenetic results by traditional calculational methods required 3,840 computational hours on the CIPRES supercomputer, publicly available at http://www.phylo.org/sub_sections/portal. Alternatively to the examples considered in section 2, this web interface may serve аs an emerging means of taxonomic analysis that uses “big” molecular genetic data and is based on traditional methods invoking, in this case, high-power computational resources.
The principal results of Hug et al. are illustrated in a very general way in (Figure 3). This scheme represents sufficiently accurately the relative locations of the nodes and branches limiting the three major domains of life (shown by different lines and dashes) in the much more detailed authors’ illustration. It also shows the set of branches, first introduced by Hug et al. (2016), which is called “candidate phyla radiation” (CPR). The evident lower phylogenetic diversity of eukaryotes has been associated with their comparatively recent evolution. The CPR set of branches is closest to the domain of Bacteria and combines prokaryotic groups for which no culturable members have been reported but which have been found to exist and contribute to biodiversity in metagenomic studies. The other nodes and branches within the four major sets in Fig. 3 were also taken from Hug et al. (2016). While these were taken arbitrarily, the relative lengths of the branches and the borders between the sets of branches were preserved.
The black circles indicate that a given taxonomic group comprises nonculturable organisms. Such organisms are absent among eukaryotes but make up 100% in the CPR set of branches. However, they also occur in considerable quantities in both prokaryotic domains (Bacteria and Archaea) and are found in them in clear-cut, fairly compact clusters in the renewed tree of life (Hug et al., 2016). Another distinctive feature of the taxons containing nonculturable organisms is that the size of the genomes of these organisms is relatively small and that they lack metabolic functions such as the full Krebs cycle, the respiratory chain, and the ability to synthesize nucleotides and amino acids. This may be due to their fairly possible symbiotic lifestyle (Hug et al., 2016), in which they transfer some of their original metabolic potential to their symbiotic partners.

Fig. 3 - Basic scheme for the renewed tree of life [from Hug et al. (2016)]. See text for details
Hug et al. noted that in many respects, the general view of the renewed tree of life (Figure 3) agrees with the tree calculated with traditional data on 16S/18S rRNA gene sequences. To this, it could be added that the nonculturable bacteria shown in the animated figure at http://www.ezbiocloud.net/ezgenome/status (Figure 2), obtained from independent data, not only form fairly isolated clusters but also occur in other analogous portions of the phylogenetic construct shown at that website.
However, the tree of life (Hug et al., 2016) provides the first quantitative demonstration that prokaryotes (mostly bacteria) make the dominant contribution to biodiversity on our planet and that nonculturable organisms (highly probably symbionts) make up the most of these. Hence, bioinformatic studies of their complete genomes are of particular significance, as they are probably the sole source of information about their metabolism and the prospects for the use of this unique microbial diversity in biotechnologies. In addition, Hug et al.’s results strongly suggest once again that the establishment of symbioses with redistribution of vital functions between partners (cooperation for joint survival) is probably the optimal and evolutionary justified form of life.
4. Origin and evolution of life as a cosmological phenomenon
Discussing the phylogenetic interrelations between organisms and the evolutionary history of life inevitably brings up the question of the origin of life. However, this question has for a long time been “factored out” of evolutionary publications, probably owing to its extreme complexity from a biological, a general scientific, and a philosophical standpoint, as well as owing to the shortage of appropriate knowledge. Only the mid-1980s saw an increase in the activity and determination of scientists dealing with this problem, also in connection with the more general cosmological questions of the origin and evolution of the entire universe (see, e.g., Koonin, 2012; Linde, 2014; Susskind, 2006; Vilenkin, 2006).
Since that time, a vast literature has accumulated in which paleontologists, astrobiologists, biochemists, geneticists, and others have discussed data on panspermia - the extraterrestrial origin of life and the migration of its simplest forms from space to the early Earth, with subsequent evolution following well-known laws. It has been pointed out that the initial stage in the evolution of life in one of its proposed major precellular forms - the RNA world (Gilbert, 1986) - could almost not have occurred in the period preceding the onset of bacterial cell life under Earth conditions (Spirin, 2001). With regard to the first geological manifestations of such life, the duration of this period has been estimated as being between ca. 4 and 3.9 billion years ago.
Life can be viewed as any temporally stable biological replicator that evolves through some combination of gene drift and natural selection (Koonin, 2012). From this standpoint, the time of the onset of life can be estimated as a period of emergence of elementary life forms with minimal biological complexity. In one of the presumably first works on this topic (Sharov, 2006) and in its further development (Sharov, 2010), such estimates were obtained by extrapolating the temporal dependence of the corresponding measures of biological (genetic) complexity of organisms to a “single gene.” It was stated that the elementary precellular life forms could not have originated on Earth 4 billion years ago and that the time interval from their possible emergence to modern days is actually ~10 billion years, exceeding considerably the geological age of Earth (~ 4.5 billion years). Cosmological data (Stenger, 2014) suggest that by that time, which is 3.8 billion years from the onset of our universe [the Big Bang (13.8 billion years ago)], light atoms had already formed in it (0.38 million years after the onset), as also had other elements of the periodic system, and the formation of galaxies, galactic clusters, and galactic superclusters was in the process of completion.
Thus, Sharov (2006; 2010) boldly puts the proposed initiation of life outside the limits of the solar system and extends the time of its evolution in the universe almost twofold: first outside Earth and then under terrestrial conditions. Note that the disagreement between the temporal evolution scales, a key factor of which was (and still remains) natural selection (Darwin, 1872), and the estimates of the age of the Earth was a concern for Charles Darwin himself. In his times, the age of the solar system was erroneously estimated to be tens of millions of years (Stenger, 2014). However, even the much more reliable modern cosmological estimates do not make this problem less acute, as shown by the results cited above.
Sharov (2006; 2010) also noted the complexity of the RNA world (no matter where it exists), leading to a fairly low probability of spontaneous self-assembly in it of biopolymeric structures and protobionts as precursors to well-developed cellular systems. He proposed a simpler and more realistic (in his opinion) model for abiogenetic selfdeveloping systems on the basis of coenzymes. In particular, the probability P of spontaneous emergence of DNA molecules of 400 base pairs in size was estimated as being fairly low (P = 10-126), even subject to the condition that all their chemical components are accessible simultaneously.
A deeper and more versatile analysis of the vanishingly small probability of several key steps in prebiological evolution and of ways to solve this paradox is given in Koonin (2007) and, in modified form, in Koonin (2012). The possibilities of spontaneous emergence of the major structural elements of living matter - chemical evolution (Koonin, 2012) and cellular and supracellular evolution under specific conditions of open systems - are beyond question and have been corroborated by convincing laboratory experiments. These include the reproduction in vitro of spontaneous emergence of amino acids (Johnson et al., 2008) and nucleobase precursors (Parker et al., 2015), the long-term evolutionary microbiological experiment described at http://myxo.css.msu.edu/ecoli/index.html, and so on. However, detailed estimations of the probability P of the emergence, by chance, of a sufficiently realistic supramolecular translation–replication system in the observable universe yielded a result even more impressive than Sharov’s (2006): P < 10-1,018 (!) (Koonin, 2012).
To solve this paradox, Koonin (2007; 2012) invoked data from cosmology and particle physics (Linde, 2014; Stenger, 2014; Susskind, 2006; Vilenkin, 2006), which form the basis for the current views on the origin, evolution, and state of the entire universe. His reasoning is based on the multiverse hypothesis (Linde 2014; 2015) in its more detailed “many worlds in one” version (Vilenkin, 2006). This hypothesis stems from the basic inflation theory of the universe and its development in the initial stages (Linde, 2005). It ensures statistical significance 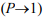 of the physical–chemical or biological processes implemented in any scenario not forbidden by conservation laws, in an infinite (Linde, 2005; 2014; 2015) or an almost infinite (Susskind, 2006) set of multiverse regions.
of the physical–chemical or biological processes implemented in any scenario not forbidden by conservation laws, in an infinite (Linde, 2005; 2014; 2015) or an almost infinite (Susskind, 2006) set of multiverse regions.
Such regions (“island universes”; Vilenkin, 2006) include the observable universe in which Homo sapiens lives. The existence of life in it, ensured by the unique values of the basic physical constants, is explained by the “anthropic principle” (Linde, 2005). According to this principle, the characteristic properties of the observable portion of the multiverse exist insofar as they ensure the existence of the observer, and they are a chance, even if optimal, set of properties among the infinite number of such sets realizable in the multiverse (Linde, 2015; Susskind, 2006; Vilenkin, 2006).
Thus, the multiverse model explains the origin of structures of any complexity that is guaranteed to be present in the infinite expanse of the multiverse (Vilenkin, 2006) and localized to the Earth under the anthropic principle (Koonin, 2007; 2012), in particular, during the realization of the above-noted “missing link” of prebiological (supramolecular) evolution. The probability of this missing link in an isolated multiverse region proved to be almost zero according to the estimates cited above. Among the multitude of possible scenarios of transition from prebiological (chemical) to biological evolution, only those are implemented that are the most probable and robust and that are compatible with the Darwinian mode of evolution of complex systems (Koonin, 2012). Already L. Boltzmann admitted the probabilistic essence of the observable universe, thinking of it as a gigantic thermal fluctuation with an entropy sufficiently low to maintain and further develop the order established in it (Boltzmann, 1898).
The cosmological aspects of this life origin and biological evolution hypothesis are associated with the inflation theory of the origin and evolution of the observable universe. Its creation and development eliminates some fundamental contradictions in the original Big Bang theory, leading to the concept of the multiverse in its eternal chaotic inflation version (Linde, 2005; 2014; 2015). The fascinating history and details of this theory merit separate consideration, as do its current state and linkage to astronomical observations. Therefore, we will only restrict ourselves to formulating some basic conclusions to support Koonin’s (2007; 2012) major idea behind the origin of life on Earth.
The theory of eternal chaotic inflation (Linde, 2005; 2015), in its most impressive segment, predicts an infinite self-replicating inflationary multiverse. In particular, it follows from this theory that if the universe contains at least one inflationary domain with suitable parameter values, it becomes to incessantly produce new inflationary domains. This process, termed eternal inflation, is maintained as a chain reaction, making the multiverse look like a fractal, as illustrated in (Figure 4) [compiled from the results in Linde (2014; 2015)].
Fig. 4 - Global structure of the chaotic self-replicating multiverse. The different gray hues symbolize “mutations” in the physics laws in the domains, as compared to the parent island universes.
Adapted from Linde (2014; 2015)
The “mutations” noted in the (Figure 4) legend may not only affect the sets of low-energy physics laws that operate in each domain (island universe) but may also change their spatiotemporal dimension (Linde, 2015). All this ensures the infinite, or almost infinite (in the string theory landscape; Susskind, 2006) statistical diversity, which is necessary for the chance (and repeated) emergence of known life forms in the multiverse under the anthropic principle (Linde, 2015). For an analogy to the Darwinian approach, see also Linde (2015) and references therein. It is appropriate in closing to quote from Stenger (2014): “Our existence on Earth is a simple matter of natural selection. With every type of planet possible in the multiverse, we naturally evolved on one with the properties needed for intelligent life.”
Список литературы
Biteen, J.S. et al. (2016) Tools for the microbiome: nano and beyond. ACS Nano, 10, 6–37.
Boltzmann, L. (1898) Vorlesungen über Gastheorie. Bd. 2. Barth, Leipzig.
Chakravorty, S. et al. (2007) A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69, 330–339.
Chen, W. et al. (2013) A comparison of methods for clustering 16S rRNA sequences into OTUs. PLOS ONE, 8, e70837.
Chun, J., Rainey, F.A. (2014) Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 64, 316–324.
Coenye T. et. al. (2005) Towards a prokaryotic genomic taxonomy. FEMS Microbiol. Rev., 29, 147–167.
Costello, M.J. et al. (2013) Can we name Earth’s species before they go extinct? Science, 339, 413–416.
Darwin, C.R. (1872) The Origin of Species by means of Natural Selection or the Preservation of Favoured Races in the Struggle for Life. Sixth Edition. John Murray, London.
De Vos, P. (2011) Multilocus sequence determination and analysis. In Rainey, F.A., Oren, A. (eds.), Taxonomy of Prokaryotes - Methods in Microbiology, Elsevier, Amsterdam, Vol. 38, pp. 385–407.
Fraser-Liggett, C.M. (2005) Insights on biology and evolution from microbial genome sequencing. Genome Res., 15, 1603–1610.
Gilbert, W. (1986) Origin of life: The RNA world. Nature, 319, 618.
Hug, L.A. et al. (2016) A new view of the tree of life. Nat. Microbiol., 1, 16048.Hug, L.A. et al. (2016) A new view of the tree of life. Nat. Microbiol., 1, 16048.
Johnson, A.P. et al. (2008) The Miller volcanic spark discharge experiment. Science, 322, 404.
Kim, M. et al. (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol., 64, 346–351.
Koonin, E.V. (2007) The cosmological model of eternal inflation and the transition from chance to biological evolution in the history of life. Biol. Dir., 2, 1–21.
Koonin, E.V. (2012) The Logic of Chance: the Nature and Origin of Biological Evolution. Pearson Education Inc., New Jersey.
Lesk, A.M. (2014) Introduction to Bioinformatics. Fourth Edition. Oxford University Press, Oxford.
Linde, A. (2005) Particle Physics and Inflationary Cosmology. Contemp. Concepts Phys., 5, 1–362.
Linde, A. (2014) Inflationary cosmology after Planck 2013. arXiv:1402.0526v2 [hep-th].
Linde, A. (2015) A brief history of the multiverse. arXiv:1512.01203v1 [hep-th]
Mora, C. et al. (2011) How many species are there on Earth and in the Ocean? PLoS Biol., 9, e1001127.
Oren, A., Garrity, G.M. (2014) Then and now: a systematic review of the systematics of prokaryotes in the last 80 years. Antonie van Leeuwenhoek, 106, 43–56.
Parker, D.S.N. et al. (2015) Gas phase synthesis of (iso)quinoline and its role in the formation of nucleobases in the interstellar medium. Astrophys. J., 803, 53–62.
Ruan, Y. et al. (2012) DACIDR: deterministic annealed clustering with interpolative dimension reduction using a large collection of 16S rRNA sequences. In BCB’12. Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, ACM New York, New York, pp. 329–336.
Sharov, A.A. (2006) Genome increase as a clock for the origin and evolution of life. Biol. Dir., 1, 1–10.
Sharov, A.A. (2010) Genetic gradualism and the extraterrestrial origin of life. J. Cosmol., 5, 833–842.
Spirin, A.S. (2001) Protein biosynthesis, the RNA world, and the origin of life. Herald Russ. Acad. Sci., 71, 146–153.
Stenger, V.J. (2014) God and the Multiverse: Humanity's Expanding View of the Cosmos. Prometheus Books, New York.
Susskind, L. (2006) The Cosmic Landscape: String Theory and the Illusion of Intelligent Design. Little, Brown, New York.
The Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature, 486, 207–214.
Tindall, B.J. et al. (2010) Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol., 60, 249–266.
Vilenkin, A. (2006) Many Worlds in One. The Search for Other Universes. Hill and Wang, New York.Vilenkin, A. (2006) Many Worlds in One. The Search for Other Universes. Hill and Wang, New York.
Woese, C.R. (2002) On the evolution of cells. PNAS, 99, 8742–8747.
Woese, C.R. et al. (1990) Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. PNAS, 87, 4576–4579.