МОДИФИКАЦИЯ ТРАНСКРИПЦИОННОГО ФАКТОРА ДЛЯ GLYCINE MAX С ЦЕЛЬЮ ПОВЫШЕНИЯ АФФИННОСТИ К CAAT-МОТИВУ IN SILICO

DOI: https://www.doi.org/10.18454/jbg.2023.20.2.001
Timkin P.D. 1 *, Penzin A.A. 2
1, 2 All-Russian Research Institute of soybean, Blagoveshchensk, Russia
* Corresponding author (tpd[at]vniisoi.ru)
Received: 16.01.2023; Accepted: 07.03.2023; Published: 26.05.2023
MODIFICATION OF THE TRANSCRIPTION FACTOR FOR GLYCINE MAX TO INCREASE AFFINITY TO THE IN SILICO CAAT-MOTIF
Research article
Abstract
Breeding new varieties of agricultural crops that are resistant to aggressive environmental conditions without reducing productivity is an important and urgent task. Modern methods of genetics and breeding make it possible to cross varieties in order to develop useful qualities and consolidate them in the population, based on genome mapping data with already annotated traits and genes. The literature offers various markers that are associated with the desired properties, such as microsatellite DNA regions (SSRs) and single nucleotide polymorphisms (SNPs), or are its direct initiator. Quantitative trait loci (QTLs) and transcription factors (TFs) are considered as initiators. If QTLs are required to increase the level of expression, then TFs are one of the key mechanisms for solving this problem. The transcription factor NFYC4 annotated in Arabidopsis thaliana plays an important role in the regulation of the plant's immune response to biotic stress and to the increase in the total protein level. For Glycine max, a homologous analogue has been presented that is responsible for the same functions. At this stage, this protein has an index of I1KC24. An increase in the expression of target genes for this protein is an important task. Protein engineering makes it possible to supplement the process of creating new stress-resistant varieties, due to additional modifications of the identified genes that are responsible for the expression of the necessary properties. In this work, one of the strategic approaches is proposed, which consists in increasing the affinity of a transcription factor for its regulatory region in DNA in silico in order to increase expression. An increase in affinity for the CAAT motif was achieved due to the introduction of multiple amino acid substitutions at the binding site for aspartic acid.
Keywords: Glycine max, I1KC24, NFYC4, affinity for DNA, in silico.
Тимкин П.Д. 1 *, Пензин А.А. 2
1, 2 Всероссийский научно-исследовательский институт сои, Благовещенск, Россия
* Корреспондирующий автор (tpd[at]vniisoi.ru)
Получена: 16.01.2023; Доработана: 07.03.2023; Опубликована: 26.05.2023
МОДИФИКАЦИЯ ТРАНСКРИПЦИОННОГО ФАКТОРА ДЛЯ GLYCINE MAX С ЦЕЛЬЮ ПОВЫШЕНИЯ АФФИННОСТИ К CAAT-МОТИВУ IN SILICO
Научная статья
Аннотация
Выведение новых сортов сельскохозяйственных культур, устойчивых к агрессивным условиям окружающей среды без снижения продуктивности является важной и актуальной задачей. Современные методы генетики и селекции позволяют скрещивать сорта сельскохозяйственных культур с целью определения полезных качеств и их закрепления в популяции, основываясь на данных картирования геномов с уже аннотированными признаками и генами. В литературе предлагаются разные маркеры, которые ассоциированы с нужными свойствами. Такими маркерами могут служить микросателлитные участки ДНК(SSRs) и однонуклеотидные полиморфизмы (SNPs). Также некоторые маркеры могут быть непосредственным инициатором нужного признака. Одними из таких инициаторов считают локусы количественных признаков (QTLs) и транскрипционные факторы (TFs). Если от QTLs требуется повышение уровня экспрессии, то к TFs в качестве одного из подходов можно попробовать увеличить его сродство. Транскрипционному фактору NFYC4, аннотированному у Arabidopsis thaliana, отводится важная роль в регуляции иммунного ответа растений на биотический стресс и на повышение общего уровня белка. Для Glycine max был представлен гомологичный аналог, отвечающий за те же функции. На данном этапе этот белок имеет идентификационный номер по базе данных Uniprot I1KC24. Повышение экспрессии генов мишеней для этого белка является важной задачей. Белковая инженерия позволяет дополнить процесс создания новых устойчивых к стрессам сортов, за счет дополнительных модификаций выявленных генов, которые отвечают за экспрессию необходимых характеристик. В данной работе предлагается один из стратегических подходов, состоящий в увеличение сродства транскрипционного фактора к его регуляторной области в ДНК in silico, с целью повышения экспрессии. Увеличение аффинности к CAAT-мотиву удалось добиться благодаря внесению множественных точечных замен аминокислот в сайте связывания на аспарагиновую кислоту.
Ключевые слова: Glycine max, I1KC24, NFYC4, аффинность к ДНК, in silico.
1. Introduction
The regulation of gene expression is one of the most important approaches in the work of breeders and geneticists, due to the complexity and high cost of establishing relationships between agricultural traits and the genes encoding them. However, work in this direction is moving forward and researchers have noted a number of markers that are associated with such traits as increased protein content or adaptation to biotic and abiotic stresses. One of these markers is QQS, for which regulatory mechanisms have been discovered and a statistical pattern has been confirmed in an increase in its expression and an increase in resistance to viruses, nematodes, a number of pathogenic bacteria, and an increase in the protein content in seeds. The transcription factor NFYC4 and, accordingly, its homologue in Glycine max are recognized as such a regulatory mechanism [1].
Transcription factors (TFs) are specialized proteins that can coordinate the expression, that is, the realization of a gene in the form of transcript production in certain cells. These proteins act as a control panel of the genome, with which you can adjust the regulation of transcription and directly form the phenotypes of the body, inducing cell survival or death [2].
NFYC4 is one of the transcription factor complex subunits required for transcription activation through binding to its specific site. Proteins of this family are expressed in almost all eukaryotes. This subunit in the complex plays the role of recognition and binding to the sequence of nucleotides in the gene that form the CAAT motif. Proteins of this family play a positive role in increasing protein content, seed germination, flowering, photomorphogenesis [3], [4], [5], [6].
CAAT motif (CCAAT-box, CAT-box) is a specific nucleotide sequence in a gene that forms a binding site for transcription factors in the promoter region. This motif is highly conserved and is usually located 60-100 bp from the transcription start site. The researchers note the great importance of these binding sites for the regulation of expression, it is also noted that a change in these motifs leads to transcriptional disruption [7].
Increasing the affinity of transcription factors for their targets will increase gene expression. Such modifications can be made using the methods and approaches of protein engineering.
This paper shows the experience of using the in silico site-directed mutagenesis approach to increase the affinity of a transcription factor for the CAAT motif.
2. Methods
1. Prediction and selection of candidates
The search for homologues was carried out in the Uniprot protein database (https://www.uniprot.org/) using the BLAST algorithm. The reference protein was the transcription factor NFYC4 from Arabidopsis thaliana, which, based on a review of the literature, was selected as the closest to similar proteins in soybeans, the selection criterion was a high level of similarity for homologues in Glycine max, due to which one can judge a similar function [8].
2. Protein modification and active site identification
According to literature sources, aspartic amino acid favorably affects binding to nucleic acids. Therefore, in the region of the primary sequence of the protein, which was predicted as a molecular pocket, multiple substitutions of native amino acids for aspartic acid were made [9]. The detection of the molecular pocket was carried out automatically at the time of modeling the interaction with DNA on the HDOCK server (http://hdock.phys.hust.edu.cn/).
3. 3D alignment of molecules
To confirm the close topology between the native protein and its modified variant, three-dimensional alignment was performed using the jFATCAT(rigid) algorithm in the RCSB Protein Data Bank web server toolkit (https://www.rcsb.org/alignment). Based on the measurement of RMSD and TM-score, conclusions were drawn about related conformational packing. This method was used to reject those candidates whose tertiary structure is not similar to the original version, due to the possible change in the coordinates of the molecular pockets for both the DNA-binding domain and the protein-binding domains necessary for the formation of a complex with other subunits. In this work, those biomolecules whose RMSD values are less than 10 and TM-score more than 0.5 were chosen as optimal metrics. 3D models of both proteins were modeled using the AlphaFold 2.0 machine learning service on the google collaborator platform (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/AlphaFold2_advanced.ipynb#scrollTo=pc5 mbsX9PZC), the source code was taken from the GitHub repository (https://github.com/sokrypton/ColabFold/blob/main/beta/AlphaFold2_advanced.ipynb), by adding primary sequences to the server.
4. Intermolecular docking
Molecular interactions between the transcription factor and DNA were modeled using the HDOCK web server. As input data for both biomolecules perceived as receptors, we used .pdb files containing the coordinates of the location of atoms in three-dimensional space. DNA sequence data were presented as primary nucleotide sequence (CCAAT)3. The interaction strength was assessed according to the Confidence score and Docking score criteria. The docking score gives an assessment of intermolecular interactions, and the more negative this indicator is, the more likely the formation of a protein-ligand complex will be. Confidence score – an indicator of reliability. Considering that protein/DNA complexes in PDBs typically have a docking score of about -200 or higher, the server empirically determines a confidence score dependent on the docking score, the probability of two molecules binding was determined by formula (1). All predicted complexes of the modified protein and DNA were visualized using the PyMOL software package.
|
Сonfidence score = 1.0/[1.0+e0.02*(Docking Score+150)] |
(1) |
When the confidence index is greater than 0.7, two molecules are more likely to bind; when the confidence index is between 0.5 and 0.7, two molecules can be linked; when the confidence index is below 0.5, the two molecules are unlikely to bind. Based on the listed metrics, two groups of results were evaluated, 10 complexes each.
3. Results and Discussion
The use of the BLAST algorithm in the Uniprot database made it possible to identify the most similar homologue for NFYC4. This homologue is a predicted protein with a similarity of 75.22% and an annotation index of 2/5. This biomolecule is the Histone domain-containing protein, which is listed in the database as I1KC24. However, such algorithms, in order to predict the function of a protein by homology, should be treated with caution, due to the lack of confirmed evidence of the relationship between the percentage of identity and similar function, and proteins that are not always similar in the primary sequence perform a similar function [10]. But in this case, according to the annotation given in the database, this protein is a transcription activator and has the ability to bind to DNA. These annotations confirm the results of the search algorithm. Modifications were made in those regions that the HDOCK web server determined to be DNA binding. Many iterations were made, resulting in the most successful variant, which showed a close topological fold with the original protein (figure 1). A successful candidate contains eight amino acid substitutions at the DNA binding sites.

Fig. 1 – Linear alignment of native protein (NATIVEPROT) and modified analog (MODPROT), unshaded purple amino acids in MODPROT indicate sites where replacements have been made
Three-dimensional alignment scores RMSD – 8.41, TM-score – 0.51, with primary sequence identity of 85%. Based on these data, we can talk about the preservation of the topological stacking, despite the rather high RMSD index, which characterizes the standard deviation of atoms in space. Nevertheless, the modified protein even visually retains the original topology (figure 2); based on all the listed metrics, it is possible to draw conclusions about the preservation of protein functions after its modifications.
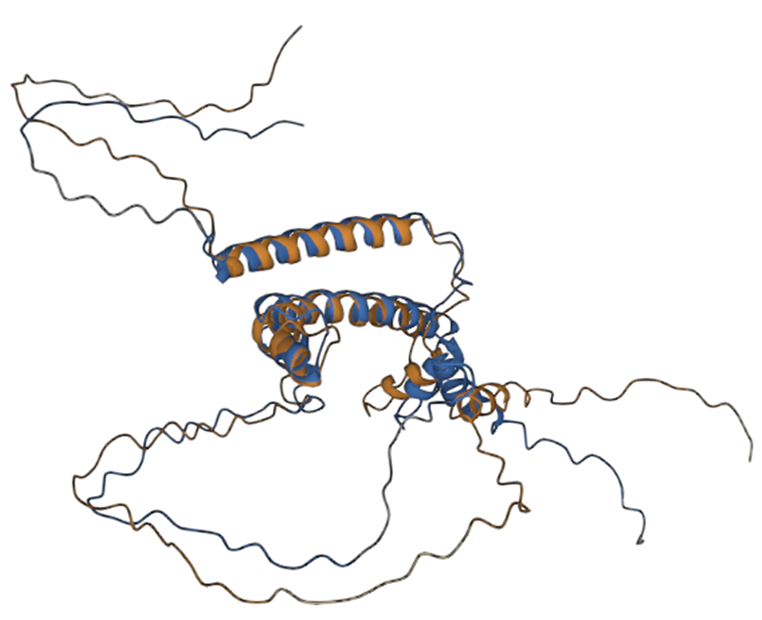
Fig. 2 – 3D alignment of native protein (blue) and modified (brown)
The results of intermolecular docking show an increase in the main parameters of the modified protein compared to the native one, characterizing the strength of interaction (table 1). The modified protein has a Confidence score in 8 out of 10 models, which characterize a high probability of interaction, compared to the native protein, where the same parameter is noted in only 1 out of 10 models. Comparing the numerical indicators of the models with each other, an increase in binding strength and probability of interaction.
Table 1 – Interaction indicators for different models of native and modified protein
|
Native protein |
Model number |
Docking score |
Confidence score |
Modified protein |
Model number |
Docking score |
Confidence score |
|
1 |
-195.78 |
0.7141 |
1 |
-230.49 |
0.8334 |
||
|
2 |
-188.20 |
0.6822 |
2 |
-200.62 |
0.7335 |
||
|
3 |
-182.02 |
0.6548 |
3 |
-199.38 |
0.7286 |
||
|
4 |
-180.62 |
0.6485 |
4 |
-198.52 |
0.7252 |
||
|
5 |
-180.11 |
0.6462 |
5 |
-198.38 |
0.7246 |
||
|
6 |
-178.44 |
0.6385 |
6 |
-193.51 |
0.7048 |
||
|
7 |
-177.66 |
0.6349 |
7 |
-192.95 |
0.7025 |
||
|
8 |
-177.54 |
0.6343 |
8 |
-192.51 |
0.7006 |
||
|
9 |
-174.02 |
0.6178 |
9 |
-191.37 |
0.6958 |
||
|
10 |
-173.07 |
0.6133 |
10 |
-191.29 |
0.6955 |
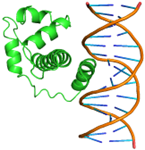
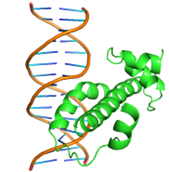
Substitutions of amino acid residues led not only to a change in the binding parameters, but also to a change in the positions of the amino acids involved in the binding (table 2). For most models, the general character of the distribution of amino acids over domains tends to be conservative and lack large differences, which indicates a change in the conformation of the DNA itself near the receptor, which was perceived by the software as a ligand. Initially predicted site-binding amino acids in the native protein located in positions: 98, 101, 102, 105, 104, 105, 106, 108, 109, 130, 134, 139, 140, 142, 143, 146, 147, coincided with the majority created models, which indicates the specificity of these sites for binding, due to their topology.
Table 2 – Models of complexes for a modified protein and amino acids involved in DNA binding
|
Model number |
Protein/DNA complex |
Amino acids bound to nucleotides |
Model number |
Protein/DNA complex |
Amino acids bound to nucleotides |
|
1 |
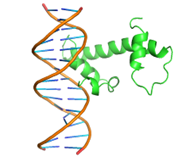 |
GLN47, GLN48, LEU50, GLN51, MET52, TRP54,SER55,TYR56, GLN59, HIS63, GLU108, ARG112, LEU115, THR135, ARG136, THR137, ASP138, ILE139 |
6 |
 |
GLN44, GLN 47, GLN48A, ASP49, LEU50, GLN51, MET52, TRP54, SER55, TYR56, GLN59, GLU60, HIS63, ARG112, ARG136, THR137, ASP138 |
|
2 |
|
GLN47, LEU50, GLN51,PHE53, TRP54, SER55, GLN57, ARG58, LEU104, GLU108, ARG112, LEU115, HIS116, GLU119, ASP120,LYS121, ASN128, ALA131, ALA132, THR135, ARG136, THR137, ASP138, ILE139, PHE140, ARG149 |
7 |
 |
GLN44, GLN48, ASP49, LEU50, GLN51, MET52, TRP54, SER55, TYR56, ARG58, GLN59, GLU60 HIS63, VAL64 |
|
3 |
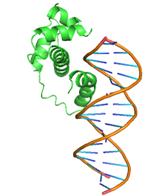 |
GLN47, GLN48, LEU50, GLN51, MET52, TRP54, SER55, TYR56, GLN59, HIS63, GLU108, ARG112, LEU115, ARG136, THR137, ASP138, ILE139 |
8 |
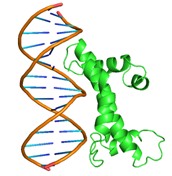 |
GLN44, GLN45, GLN46, ASP49, LEU50, MET52, PHE53, TYR56, GLN59, GLU60, HIS63, VAL64 ASN65, GLN71, LYS100, LEU107, THR110, ILE111, ASP114, GLU118, GLU119, LYS121, ARG123 |
|
4 |
 |
GLN44, GLN46, GLN48, ASP49, GLN51, MET52, PHE53, SER55, TYR56, GLN59, GLU60, VAL64, ASN65, ASP66, PHE67, LYS68, ASN69, HIS70, GLN71, LEU72, PRO73, LYS100, GLU103, LEU104, LEU107, THR110, ASP114 |
9 |
 |
GLN44, GLN47, GLN48, ASP49, LEU50, GLN51, MET52, TRP54, SER55, TYR56, GLN59, GLU108, ARG112, LEU115, ARG136, THR137, ASP138 |
|
5 |
 |
GLN44, GLN47, GLN48, ASP49, LEU50, GLN51, MET52, PHE53, TRP54, SER55, TYR56, GLN59, HIS63, GLU108, ARG112, LEU115, THR135, ARG136, THR137, ASP138, ILE139 |
10 |
 |
GLN44, GLN45, GLN48, ASP49, GLN51, MET52, TRP 54, SER55, TYR56, ARG58, GLN59, GLU60, HIS63, ARG112,THR137 |
4. Conclusion
As a result of the study, it was possible to increase the affinity of a transcription factor for its specific region of the gene by replacing some amino acids in the putative molecular pocket. It is possible to confirm the concept of the positive effect of aspartic acid, if not on the direct participation in binding, then the creation of favorable conditions for the binding of nearby amino acids. These data may further assist in the development of protocols for in vitro and in vivo modifications to improve the beneficial properties of cultivated soybeans. Understanding the mechanisms of regulation of gene expression and possible approaches to manipulation and changes in the efficiency of nucleic acids will allow the creation of plant varieties with already specified properties and parameters.
The obtained results of chemoinformatic prediction should be verified in vitro using chip-seq or cryoelectron microscopy technologies. The use of these methods will directly confirm the accuracy of the predictive model proposed in the work. An increase in the affinity of transcription factors for target motifs may open up a new strategy in biotechnology in the future.
|
Conflict of Interest |
Конфликт интересов |
|
None declared. |
Не указан. |
Список литературы
Qi M. QQS orphan gene and its interactor NF-YC4 reduce susceptibility to pathogens and pests / M. Qi, W. Zheng, X. Zhao [et al.] // Plant Biotechnol J. 2019 Jan; 17(1): 252-263. — DOI: 10.1111/pbi.12961. — Epub 2018 Jul 6. PMID: 29878511; PMCID: PMC6330549.
Francois M. Modulating transcription factor activity: Interfering with protein-protein interaction networks / M. Francois, P. Donovan, F. Fontaine // Semin Cell Dev Biol. 2020 Mar; 99: 12-19. — DOI: 10.1016/j.semcdb.2018.07.019. — Epub 2018 Sep 13. PMID: 30172762.
Tang Y. Arabidopsis NF-YCs Mediate the Light-Controlled Hypocotyl Elongation via Modulating Histone Acetylation / Y. Tang, X. Liu, X. Liu [et al.] // Mol Plant. 2017 Feb 13;10(2): 260-273. — DOI: 10.1016/j.molp.2016.11.007. — Epub 2016 Nov 19. PMID: 27876642.
Li L. QQS orphan gene regulates carbon and nitrogen partitioning across species via NF-YC interactions / L. Li, W. Zheng, Y. Zhu [et al.] // Proc Natl Acad Sci USA. 2015 Nov 24;112(47): 14734-9. — DOI: 10.1073/pnas.1514670112. — Epub 2015 Nov 9. PMID: 26554020; PMCID: PMC4664325.
Liu X. The NF-YC-RGL2 module integrates GA and ABA signalling to regulate seed germination in Arabidopsis / X. Liu, P. Hu, M. Huang [et al.] // Nat Commun. 2016 Sep 14;7: 12768. — DOI: 10.1038/ncomms12768. — PMID: 27624486; PMCID: PMC5027291.
Kumimoto R.W. NF-YC3, NF-YC4 and NF-YC9 are required for CONSTANS-mediated, photoperiod-dependent flowering in Arabidopsis thaliana / R.W. Kumimoto, Y. Zhang, N. Siefers [et al.] // Plant J. 2010 Aug;63(3): 379-91. — DOI: 10.1111/j.1365-313X.2010.04247.x. — Epub 2010 May 6. PMID: 20487380
Bezzecchi E. NF-YA Overexpression in Lung Cancer: LUSC / E. Bezzecchi, M. Ronzio, D. Dolfini [et al.] // Genes (Basel). 2019 Nov 17; 10(11):937. — DOI: 10.3390/genes10110937. — PMID: 31744190; PMCID: PMC6895822.
Sinha S. Predicting Protein Function Using Homology-Based Methods / S. Sinha, B. Eisenhaber, A.M. Lynn; ed. by Shanker A. // Bioinformatics: Sequences, Structures, Phylogeny. — Springer, Singapore, 2018. —DOI: 10.1007/978-981-13-1562-6_13
Lin M. New insights into protein-DNA binding specificity from hydrogen bond based comparative study / M. Lin, J.T. Guo // Nucleic Acids Res. 2019 Dec 2;47(21): 11103-11113. — DOI: 10.1093/nar/gkz963. — PMID: 31665426; PMCID: PMC6868434.
Whisstock J.C. Prediction of protein function from protein sequence and structure / J.C. Whisstock, A.M. Lesk // Quarterly Reviews of Biophysics. 36 (3): 307–40. August 2003. — DOI:10.1017/S0033583503003901. — PMID 15029827. S2CID 27123114