Bridging the Gap Between Genotype and Phenotype: Integrating Genome-Wide Association Studies and Computational Structural Biology to Decipher the Mechanisms of SNP-Induced Protein Misfolding in Human Disease
Bridging the Gap Between Genotype and Phenotype: Integrating Genome-Wide Association Studies and Computational Structural Biology to Decipher the Mechanisms of SNP-Induced Protein Misfolding in Human Disease

Abstract
Genome-wide association studies (GWAS) have identified thousands of genetic loci linked to complex human diseases; however, elucidating the biological mechanisms underlying these associations remains a major challenge, commonly referred to as the genotype–phenotype gap. The development of polygenic risk scores (PRS), which integrate the cumulative effects of numerous small-effect variants into a single risk metric, has significantly advanced population-level disease prediction. Notably, PRS have been shown to identify individuals whose disease risk rivals that conferred by rare monogenic mutations. Despite their predictive power, PRS offers limited mechanistic insight, as they obscure the functional consequences of individual variants and their collective impact on molecular pathways. A common mechanistic thread among many disease-associated non-synonymous single nucleotide polymorphisms (nsSNPs) is the perturbation of protein folding thermodynamics, resulting in reduced stability, aberrant aggregation, and loss of protein function. This review presents an integrative framework that bridges GWAS discoveries with computational structural biology to address this gap. We outline the biophysical principles governing mutation-induced protein destabilization, drawing on well-characterized disease models such as p53, CFTR, prion proteins, and SOD1. The role of GWAS endophenotypes in uncovering candidate pathogenic nsSNPs is also discussed. Central to this review is a computational pipeline combining pathogenicity prediction, protein stability change (ΔΔG) estimation, and molecular dynamics simulations to assess structural disruption. Applications to cancer-associated genes, including RTEL1, MLH1, and NBN, alongside network-based analyses, demonstrate the translational potential of this approach. Finally, we highlight how these mechanistic insights can inform rational drug design, exemplified by in silico stabilization strategies for misfolded p53. Integrating population genetics with atomistic modeling offers a powerful route to transform polygenic associations into actionable biological mechanisms and therapeutic targets.
1. Introduction
1.1. The GWAS Revolution and the Challenge of Interpretation
The innovation of genome-wide association studies (GWAS) has significantly impacted our knowledge of the genetic factors that contribute to human diseases of complex nature. Essentially, GWAS have identified thousands of loci that are statistically correlated with an array of disorders from Alzheimer's disease to cancer , through the genotyping of up to millions of single nucleotide polymorphisms (SNPs) in large cohorts. The establishment of such studies has not only extensively confirmed the polygenic nature of most common diseases but also unveiled new paths of biological mechanisms , . Nevertheless, this accomplishment in discovering genetic signals has led to another challenge: the genotype-phenotype gap . Most of the variants that have been identified lie in the non-coding regions, and for causative non-synonymous SNPs (nsSNPs), even the statistical association is generally not sufficient to locate the biological mechanism. A single nucleotide polymorphism that is linked to the increased susceptibility of a disease does not indicate the manner in which it disrupts the normal cellular function, which is the key issue in the development of targeted therapies , .
Yet quantitative analyses have demonstrated that, despite these successes, GWAS signals often explain only a modest fraction of total heritability for complex traits. Individual common risk variants typically explain only ~0.02–0.3% of phenotypic variance even in large meta-analyses, and the total variance explained by all known loci frequently falls well below the heritability estimates derived from family and twin studies , . This ‘missing heritability’ problem highlights that statistical association alone is not sufficient to understand how specific variants perturb molecular processes . Adding complexity, many lead SNPs fall within non-coding regions or in LD blocks spanning multiple genes, further obscuring identification of the true causal variant and its molecular mechanism , . Such challenges motivate integrative approaches that combine GWAS with functional genomics and structural biology .
1.2. The Protein Misfolding Paradigm: A Unifying Mechanism
A significant and uniting mode of nsSNP pathogenicity is the disruption of protein stability and folding , . For most proteins, function is not necessarily a matter of the exact chemistry of an active site but of the protein's capacity to form and sustain a specific, thermodynamically stable three-dimensional shape. A vast number of disease-causing nsSNPs have their impact not by directly modifying active site amino acids but by destabilizing the native protein conformation , . Such destabilization lowers the folding free energy of the protein (ΔG), raising the fraction of misfolded, aggregation-inducing intermediates. Newly changed shapes are usually recognized to the cell quality control systems (e.g. the ubiquitin-proteasome pathway), that changes/fail and thus leads to reduction of the normal function , . However, they may also coalesce into the nucleation of toxic oligomers, or into the insoluble aggregates that not only physically block the cell but also become a source of toxic gain-of-function , . This illustration is a good point of those mutations that are randomly distributed along a protein amino acid sequence but still may lead to one common pathological effect proteostasis dis-balance , .
The Protein Misfolding Mechanism
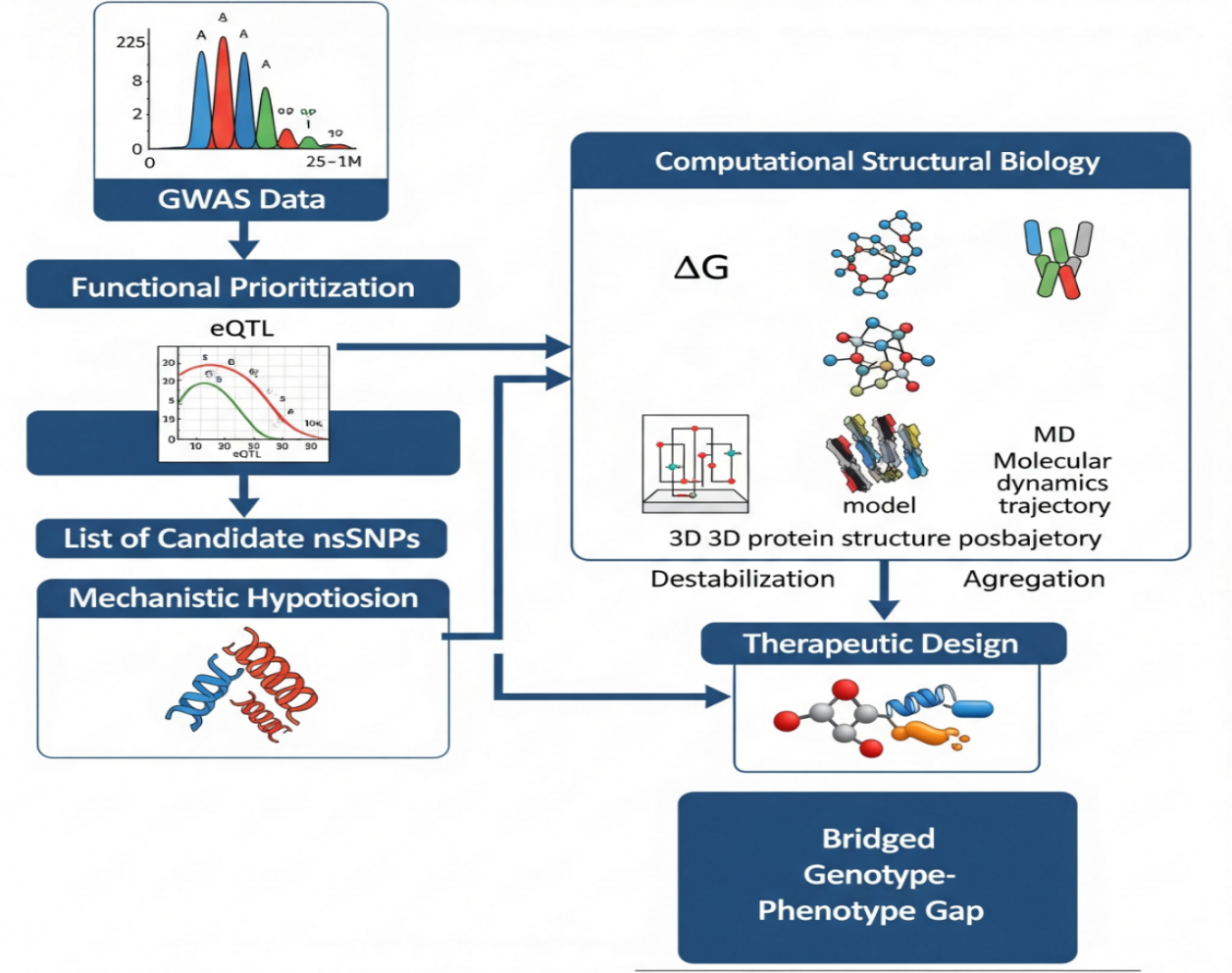
The main purpose of this review is to persuade the readers that bridging the genotype-phnotype gap is a challenge that requires the integration of GWAS with computational and experimental structural biology on a systematic basis. The authors here put forward a staged plan that uses as input the outcomes of GWAS—ranked lists of nsSNPs—and goes on with thorough in silico operations for the production of accessible physicochemical conjectures around protein stability. This review shall:
1. Detail the biophysical bases of protein misfolding diseases.
2. Describe the present GWAS techniques for setting up candidate lists of nsSNPs.
3. Outline a computational workflow for the study of nsSNPs by step-by-step explanation.
4. Give the research instances of employing this pipeline in cancer and neurodegenerative disease genes.
5. Consider the viewpoint of treatment and the further development of this combining approach , .
2. The Fundamental Principles of Protein Misfolding in Disease
2.1. The Protein Folding Energy Landscape
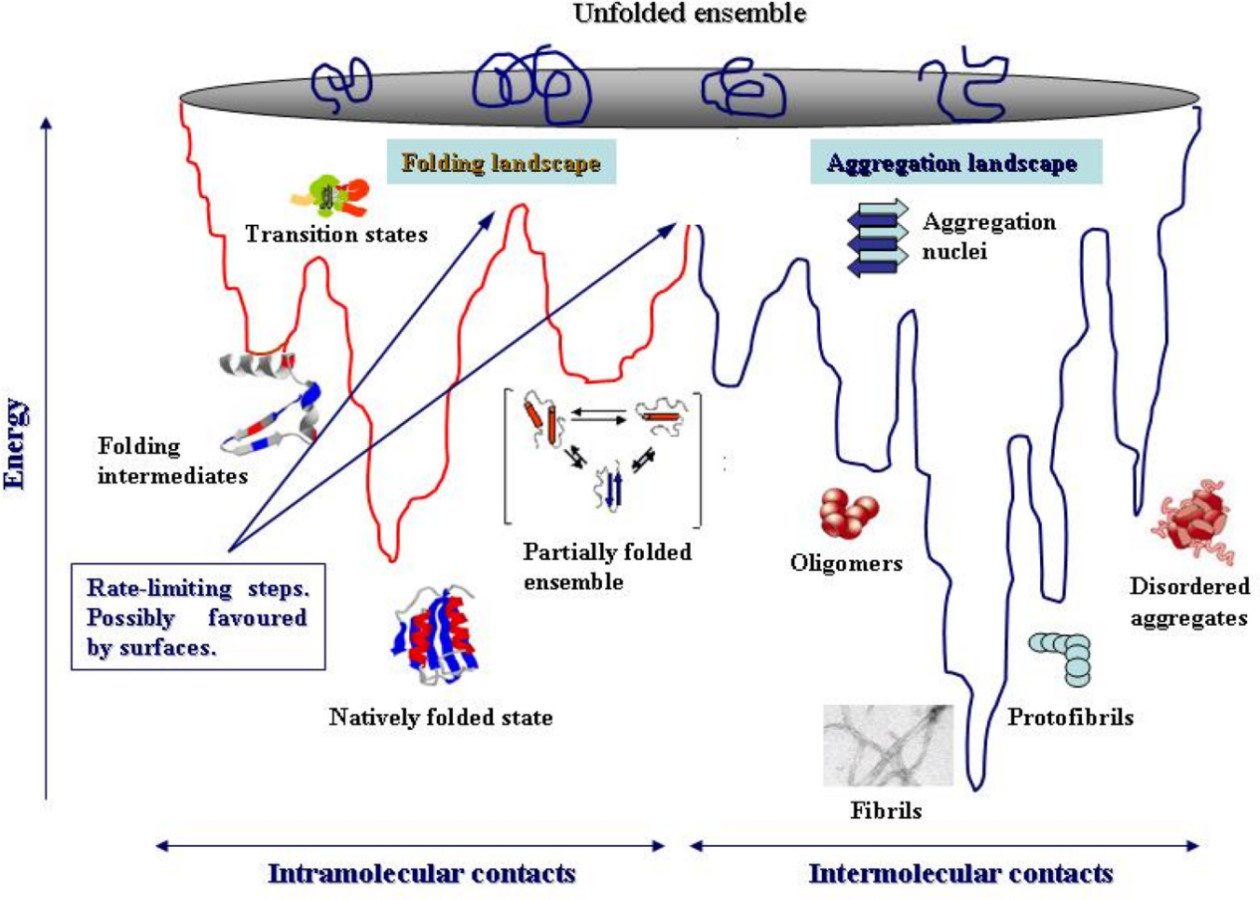
Proteins fold across a vast energy landscape, conceptually visualized as a funnel (Figure 2). The wide top represents the ensemble of innumerable unstructured conformations with high entropy and high free energy. The narrow bottom represents the unique, biologically active native state, which resides at a global free energy minimum. The depth of this minimum represents the thermodynamic stability of the folded protein, quantified by the Gibbs free energy of folding, ΔG = G_folded - G_unfolded. A large, negative ΔG indicates a stable protein. The rugged sides of the funnel represent kinetic barriers between metastable intermediate states. The folding process is a biased random search across this landscape, guided by the principle of energy minimization. Mutations can change this landscape in two main ways: (1) by raising the energy of the native state, which makes it shallower and less stable, or (2) by lowering the energy of off-pathway intermediates, which creates kinetic traps that lead to misfolded aggregates .
The Protein Folding Energy
A single amino acid substitution can destabilize a protein through several distinct mechanisms, often quantified by the change in folding free energy, ΔΔG = ΔG_mutant - ΔG_wild-type. A negative ΔΔG indicates a destabilizing mutation. Destabilizing Interactions: Inserting a charged or polar residue into the hydrophobic core can be highly destabilizing due to the high energetic cost of desolvating that group. Similarly, introducing a large residue can cause steric clashes that distort the backbone. Disruption of Hydrophobic Core Packing: The hydrophobic effect is a major driving force for folding. As an example, if you make a hydrophobic substitution in the core with a smaller residue, i.e., mutate Ile to Val, this will cause the creation of cavities that have fewer van der Waals interactions than previously and will thus destabilize the core. Disruption of Stabilizing Interactions: The extent of disruption by a point mutation is so radical that it can disassemble the particular stabilizing interactions that are the basis of the native folded structure support, Salt bridges (e.g., exchanging Arg for Ser in a pair with Asp) being one group, while hydrogen bonds, or π-stacking interactions between aromatic residues could be others.
Reduction of Protein Stability: the changes can increase the flexibility of loops or secondary structure elements, leading to a decrease in the unfolding energy barrier and thereby causing an increase in the thermodynamic favorability of the unfolded states. Experimental and bioinformatic analyses uniformly point to the emerging view that many disease-causing missense mutations are only mildly to moderately destabilizing in thermodynamic terms but have disproportionate biological consequences. Typical pathogenic nsSNPs cause folding free energy changes (ΔΔG) in the range of +1 to +5 kcal/mol, sufficient to push marginally stable proteins into regimes of partial unfolding and aggregation. For instance, in p53, hotspot tumor mutations such as R175H, R249S, and Y220C lower the melting temperature (Tₘ) of the DNA-binding domain by 5–10°C and shift the folding equilibrium towards unfolded and aggregation-prone species. Similarly, in SOD1, ALS-associated variants weaken the dimer interface, increasing the monomer population and exposing hydrophobic surfaces that seed toxic aggregates; destabilization in the range of 1.5–3 kcal/mol correlates with aggregation propensity and disease severity. These examples illustrate how limited changes in free energy may significantly alter the topology of the energy landscape and balance of proteostasis .
2.3. Experimental Foundations: Lessons from Key Proteins
2.3.1. p53: A Prototype for Misfolding in Cancer
Among all cancer-causing genes, it is the gene that codes for the tumor suppressor protein p53, which in turn controls oncogenes, that is altered most frequently. It is so often mentioned as a classic case of a mutation causing misfolding. Accordingly it has indicated that the mechanism for loss of function of p53 protein through various cancer mutations is the destabilization of the overall structure and not only the active site.
Key Findings: these researchers after investigating the structure of p53 that binds DNA under standard body conditions (37 C) reached a conclusion that the mutants located in the hotspot such as R249S and R175H:
Lowering of the melting temperature (Tm) significantly: the reduction of 5-10°C that is observed here is indicative of a large loss of thermodynamic stability.
Advantage aggregation behavior to a great extent: the mutated proteins that are destabilized proceed to the stage of fibril formation that is very close to the one observed for amyloid in vitro. Along with the residues that bind DNA unchanged, they cause a loss of function: the evidence points to the defect of the protein ascribed to its failing to keep the native fold stable rather than the direct interruption of DNA-contact sites
.Significance: this work established that a mutation's location is less important than its effect on global stability. It shifted the perspective on p53 dysfunction from a "local" to a "global" problem.
2.3.2. CFTR: Misfolding and the Path to Personalized Therapy
The tale of cystic fibrosis transmembrane conductance regulator (CFTR) is the leading success story of the study of a misfolded protein's molecular mechanism resulting in an effective, personalized, and therapeutically targeted treatment strategy.
Key Findings: the predominant mutation, ΔF508, causes cystic fibrosis not by destroying ion channel function but by causing the protein to be recognized as misfolded by the endoplasmic reticulum (ER) quality control machinery. The mutation disrupts the first nucleotide-binding domain (NBD1) of the protein, consequently, the protein cannot be normally folded. The aberrant protein, which is being re-exported from the ER, is marked with ubiquitin, thus, it is degraded by the proteasome via ER-associated degradation (ERAD). Due to this, very low levels of CFTR are present in the plasma membrane, which means that the loss of chloride conductance occurs.
Therapeutic Translation: this mechanism that was understood from the valve-function has resulted in the following innovations:
CFTR correctors (e.g., lumacaftor, tezacaftor, elexacaftor): a small-molecule entity that, as a pharmacological chaperone, attaches and stabilizes the newly made ΔF508-CFTR protein, hence, the folding and the release to the cell membrane are made possible.
CFTR potentiators (e.g., ivacaftor): those are the agents that, after membrane targeting, facilitate the channel-open probability of CFTR, thus the combined therapy of such kind is a remarkable example of the success of the structure-based drug design in the protein misfolding disorder field
.2.3.3. Prion Proteins: Probing Early Misfolding Events
Prion diseases constitute lethal neurodegenerative ailments with the unique hallmark of an abnormal conformational change of the normal cellular prion protein (PrP^C) to a pathogenic aggregated isoform (PrP^Sc). Giachin et al. utilized solution-state NMR spectroscopy to investigate the initial atomic level structural perturbations caused by pathogenic mutations. (e.g., E200K).
Key Findings: their work revealed that disease-associated mutations do not cause large-scale structural changes in PrP^C. Instead, they induce. Changes in dynamics: Increased flexibility or rigidity in specific loops and helices.
Altered stability: a reduction in the thermodynamic stability of the C-terminal domain. Perturbed hydrogen-bonding networks: changes in key interactions that stabilize the native fold. Significance represents the minor alterations that substantially reduce the amount of energy required for the transition to the PrP^Sc state. Besides a very close and temporary perspective of the "zero point" of misfolding, NMR revealed the single-point mutation mechanism that turns the protein into a lethal conformational change while still preserving its unchanged structure .
2.3.4. SOD1: Misfolding and Aggregation in ALS
Pal et al. investigated how mutations in the conserved regions of SOD1 affect the severity of the disease and the treatment outcome from the standpoint of a drug-targeted approach. It is because several mutations in the gene that produce the copper-zinc superoxide dismutase (SOD1) enzyme have been found to be highly correlated with the onset of Amyotrophic lateral sclerosis (ALS).
Key Findings: the SOD1 protein is dimer that is made up of 2 identical subunits with very strong interactions between them and low dissociation constant (Kd). Mutations like A4V and G93A.
Destroy the original dimer: these mutations enhance the dissociation constant, thus, the amount of the unstable monomers is increased.
Make hydrophobic surfaces visible: the unstable monomers expose those parts of the aggregation-prone regions that were concealed in the dimer interface.
Attract toxic aggregation: the monomers can easily combine into one type of aggregates – insoluble, cytotoxic – that are the cause of ALS pathology, thus, are the hallmarks of this disease.
Connect with severity: the degree of destabilization and aggregation propensity most times mirror the clinical severity and the rate of disease progression.
Significance: the findings of the present study have resulted in the change of the modality of SOD1-ALS therapeutic interventions from those that augment enzymatic activity to therapies that focus on the stabilization of the native dimeric state or prevention of monomeric aggregation, hence, again pointing out stabilization as the main therapeutic target
.
3. Genomic Discovery: From GWAS to Candidate Genes
3.1. The Architecture of GWAS: Identifying Risk Loci
GWAS means a non-biased, complete, and systematic study to investigate correlations between genetic changes (typically single nucleotide polymorphisms or SNPs) across the entire genome and phenotypes or illnesses. In a typical case-control GWAS, millions of SNPs are genotyped or imputed in thousands of subjects
. For the SNPs an association test is conducted (e.g., chi-squared or logistic regression) testing for a difference in allele frequencies between cases and controls. The outcomes represent a Manhattan plot, where every point is a SNP, the x-axis value represents its genome location, and the y-axis is the -log10(p-value) for the association. Places where the peaks exceed a determined genome-wide significance threshold (usually p < 5 × 10^-8) suggest loci in the genome that the implied risk variants are originating from .3.2. Beyond the Hype: The "Missing Heritability" and Endophenotypes
One of the major criticisms of GWAS is that there are heaps of 'missing heritability'—where multiple common variants combined make up only a small fraction of the total genetic variance estimated from family studies. The number of associated loci is generally large and encompasses several genes, many non-coding variants, and long-range linkage disequilibrium that complicates the identification of genes and causal variants. Endophenotypes represent a strong approach for circumventing these potential limitations. Endophenotypes are heritable, disease-relevant quantitative biochemical or physiological traits that lie between the genotype and the clinical phenotype. They are thought to be closer to the underlying biological action of genes than the complex clinical syndrome. Researchers use endophenotypes to associate genes to diseases in genome-wide association studies due to greater statistical power of these causal traits .
Beyond endophenotypes, Mendelian randomization has emerged as a powerful, complementary approach to GWAS in reaching inferences about causality and prioritizing candidate genes. MR uses naturally occurring genetic variants, usually SNPs associated with a modifiable risk factor-for example, a protein level, metabolite, or lifestyle biomarker-as instrumental variables in testing for the causal effect of such a factor on a disease outcome. This method is based on the principle that genetic alleles are randomly assigned at conception, mimicking a randomized controlled trial and largely avoiding the reverse causation and confounding biases that plague observational epidemiology. For instance, a study by Brumpton et al. applied MR to demonstrate causal effects of cardiovascular risk factors on a range of diseases. Critically, when the genetic instrument is a non-synonymous SNP (nsSNP) that directly alters the amino acid sequence of a protein, the MR inference gains profound mechanistic weight. A positive MR result in this context suggests that the variation in disease risk is causally mediated through the alteration of that specific protein's function. Consequently, genes implicated through such nsSNP-led MR analyses become high-priority candidates for the computational structural pipeline described herein. They represent not simply statistical associations but hypothesized causal levers, wherein the mechanistic hypothesis-that the nsSNP disrupts protein stability or interactions-is explicitly testable through molecular dynamics and biophysical modeling. Therefore, MR provides the necessary filter of genetic association with strong causal inference that may be directly fed into structural biology workflows to interrogate the precise atomic level of dysfunction and guide the rational design of therapeutics , .
3.3. Case Studies in Alzheimer's Disease: Leveraging Endophenotypes
3.3.1. Deming et al.: Endophenotypes and Disease Modifiers
Deming and colleagues were not based on clinical AD diagnosis for their GWAS. They went forward with GWAS on deeply phenotyped data or endophenotypes.
Main Conclusions: The researchers measured the levels of main biomarkers for Alzheimer's disease (AD) in cerebrospinal fluid (CSF) (Aβ42, t-tau, p-tau) and performed cognitive evaluations on the patients. A big success of them was to discover four signals that are associated with these endophenotypes and which had not been found in previous case-control studies.
Biological Understanding: One of the most important things was that the genes located next to those loci are very close to the atomic events that characterize the main features of the disease (e.g., amyloid and tau pathologies), immune response, and endocytic trafficking. As a result, this approach can uncover genes and pathways that are biologically strongly supported but still quite challenging to recognize in diagnosis-focused research .
3.4. Moving from Locus to Gene: Strategies for Prioritization
Identifying the risk locus is only the very beginning of the story. The next milestone, very significant, is to show the exact gene and variant that cause the problem. The works of Chen et al. and Cano-Gamez & Trynka give a very broad overview of the fundamental criteria for selecting candidate genes as well as the possible methods for functional prioritization.
Functional Annotation: Researchers can pinpoint trait-related SNPs together with functional genomic regions through exploration of easily accessible resources like RegulomeDB or HaploReg (promoters, enhancers, transcription factor binding sites, etc.).
Expression Quantitative Trait Locus (eQT L) Mapping: The most primitive thing to do is to see whether the SNP genotype has a connection with the expression levels of the genes in its neighborhood (thus, the SNP is an eQTL). Colocalization tools (e.g., COLOC) can infer a single variant responsible for both the GWAS signal and the expression signal. The gene closest to the risk locus and the SNP that is eQTL will be those of the most likely causal candidates.
Chromatin Interaction Maps (Hi-C): With the help of genome-wide chromatin interaction data, the SNP region can be investigated to find out whether it is connected to the promoter of a far-off gene via chromatin folding, even if the gene is megabytes away.
Integration with Other Omics Data: The process of layering GWAS hits with epigenomic data (ChIP-Seq for histone marks), proteomic data (pQTLs), and metabolomic data (mQTLs) is that of prioritizing and ranking candidate nsSNPs in coding regions for further detailed structural analysis [37].
4. The Computational Bridge: From nsSNPs to Mechanistic Hypotheses
Functional genomics produce a prioritized list of candidate non-synonymous single nucleotide polymorphisms (nsSNPs). The next crucial step is to figure out their atomistic mechanistic impact. A multi-step in silico pipeline that predicts structural and functional consequences is used to achieve this [38].
4.1. The In Silico Pipeline for Analyzing Pathogenic nsSNPs
A structured computational workflow is the key to a systematic evaluation of nsSNPs (Figure 3). This pipeline changes a variant of unknown significance into a hypothesis that can be tested about protein stability and function.
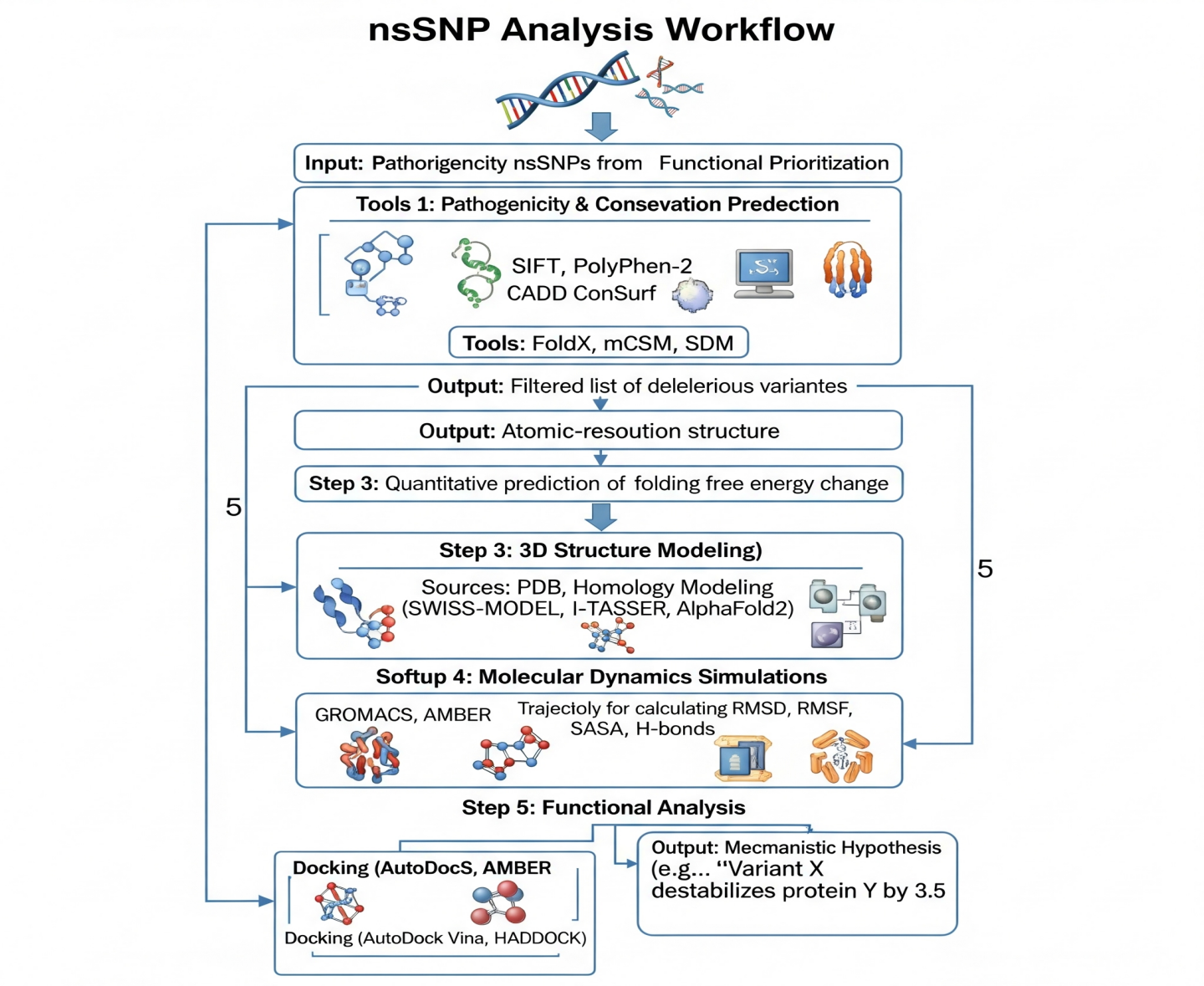
The Integrated Computational Pipeline for nsSNP Analysis
Step 1: Pathogenicity & Conservation Prediction. Tools: SIFT, PolyPhen-2, CADD for pathogenicity; ConSurf for evolutionary conservation. Output: Filtered list of deleterious variants.
Step 2: Stability Prediction (ΔΔG). Tools: FoldX, mCSM, SDM. Output: Quantitative prediction of folding free energy change.
Step 3: 3D Structure Modeling. Sources: PDB, Homology Modeling (SWISS-MODEL, I-TASSER, AlphaFold2). Output: Atomic-resolution structure.
Step 4: Molecular Dynamics Simulations. Software: GROMACS, AMBER. Output: Trajectories for calculating RMSD, RMSF, SASA, H-bonds.
Step 5: Functional Analysis. Docking (AutoDock Vina, HADDOCK) to study binding interfaces
Output: Mechanistic Hypothesis (e.g., "Variant X destabilizes protein Y by 3.5 kcal/mol, increasing flexibility in functional loop Z and disrupting binding to partner W"). In practice, the pipeline converts qualitative questions (‘Is this variant important?’) into quantitative biophysical measurements. Pathogenicity tools (SIFT, PolyPhen-2, CADD) generate probability scores of deleteriousness; stability tools (FoldX, mCSM, SDM) return ΔΔG estimates often in the range of −6 to +6 kcal/mol, where positive values indicate destabilization. These predictions are then contextualized by MD-derived observables such as shifts in average RMSD, increased RMSF in functional loops, changes in SASA of hydrophobic cores, and loss of key hydrogen bonds or salt bridges, collectively forming a mechanistic hypothesis for how an nsSNP perturbs structure and function [39], [40].
4.2. Predicting Pathogenicity and Functional Impact
The initial filter assesses whether an amino acid change is likely deleterious. SIFT (Sorting Intolerant From Tolerant): The main idea behind SIFT is to judge whether a change in protein function happens due to the change in amino acid sequence of the protein if the mutated residue is evolutionarily related to the referenced wild-type and has similar physical properties. The prediction of the effect as damaging is done when the score obtained is less than 0.05 [41].
PolyPhen-2 (Polymorphism Phenotyping version 2): Uses machine learning based on sequence, phylogenetic, and structural features to classify variants as "probably damaging," "possibly damaging," or "benign." CADD (Combined Annotation Dependent Depletion): Integrates over 60 functional annotations into a single C-score. A higher score (e.g., >20) indicates a variant is more likely to be deleterious. Conservation Analysis with ConSurf: Estimates the evolutionary conservation of amino acid positions. Residues with a high conservation score (e.g., 8–9) are the most conserved for structure/function, and changes in such positions are more likely to be the cause of a disease [42].
4.3. Predicting Energetic Consequences: Computing ΔΔG of Folding
This stage is the most significant for the evaluation of the misfolding risk, where the protein folding free energy change (ΔΔG) prediction is carried out.
FoldX: It is a straightforward process that is modeled on a real energy function. The Build Model command is responsible for synthesizing the mutant as well as calculating the energy difference for the two structures, wild-type, and mutant. ΔΔG < 0 kcal/mol is called destabilization [43].
mCSM: It creates graph-based signatures for depicting the environment of wild-type structures, and it employs machine learning for the prediction of protein stability upon mutations.
SDM (Site-Directed Mutator): An energy function that is based on statistical potentials and uses amino-acid substitution tables specific to the protein environment, which are derived from protein domain families, for scoring stability [44].
Table 1 - Summary of Key Computational Tools for nsSNP Analysis
Analysis Type | Tool Name | Methodology | Key Output |
Pathogenicity | SIFT | Sequence homology-based | Score (0.0 deleterious - 1.0 tolerated) |
PolyPhen-2 | Machine learning-based | Score (Benign, Possibly, Probably Damaging) | |
CADD | Integration of multiple annotations | C-score (Higher = more deleterious) | |
Conservation | ConSurf | Evolutionary analysis | Score (1–9, 9 most conserved) |
Stability (ΔΔG) | FoldX | Empirical force field | ΔΔG (kcal/mol) |
mCSM | Machine learning & graph-based | ΔΔG (kcal/mol) | |
SDM | Statistical potential | Stability score | |
Structure | SWISS-MODEL | Homology modeling | 3D Protein Structure |
AlphaFold2 | Deep learning (AI) | 3D Protein Structure | |
Dynamics | GROMACS | Molecular Dynamics | Trajectories (RMSD, RMSF, SASA) |
AMBER | Molecular Dynamics | Trajectories (RMSD, RMSF, SASA) | |
Docking | AutoDock Vina | Docking simulation | Binding affinity (kcal/mol) |
4.4. Modeling Structural Derangement: Homology Modeling and Molecular Dynamics
Homology Modeling: When the structure of a protein is not available experimentally, software such as SWISS-MODEL and I-TASSER create a 3D model based on a similar protein structure. The prediction by AlphaFold2 has changed the face of this domain with its near-perfect ab initio predictions.
Molecular Dynamics (MD) Simulations: GROMACS and AMBER, for instance, are the programs which physically simulate the movements of atoms, over a particular time period, hence giving the protein's dynamic behavior that static structures lack.
Root Mean Square Deviation (RMSD): A method to show the average difference in distance between atoms of a protein when compared with a standard frame. Over a mutant, a high and variable RMSD is generally related to the global structural instability. Root Mean Square Fluctuation (RMSF): The flexibility of a single residue is measured here. The increase of the flexibility (higher RMSF) in the non-catalytic region (e.g., binding loops) would imply that the function is lost.
Solvent Accessible Surface Area (SASA): The measure of the hydrophobic residues that are exposed to water. The rise of this parameter indicates the partial unfolding of the protein and the consequent exposure of the hydrophobic core, which is the main reason for the aggregation process.
Hydrogen Bond & Salt Bridge Analysis: One of the methods to measure the loss or formation of long-range interactions which contribute to protein stability [45].
4.5. Case Studies: Applying the Pipeline
4.5.1. A Network View of Stability Loss in Cancer Genes
Reza brought to light the natures of the nonsynonymous single nucleotide polymorphisms (nsSNPs) in a network of the top interconnected genes related to cancer susceptibility.
Methods & Findings: They combined protein-protein interaction (PPI) network analysis with their computational pipeline for the main result that pathogenic nsSNPs were found more than expected in hub genes and were predicted to cause a significantly larger destabilization (more negative ΔΔG) than neutral polymorphisms.
Significance: Their study linked a systems-level perspective, illustrating that mutations knocking out the central network nodes have the highest, disease-causing impact on the cell function, thus giving an explanation of their strong GWAS association [46].
4.5.2. A Comprehensive In Silico Investigation of RTEL1
Tanshee et al. and co-workers in silico extensively examined the nsSNPs of RTEL1 helicase which was identified as one of the main factors responsible for telomere maintenance.
Method: They employed sets of tools such as SIFT, PolyPhen-2, PROVEAN for phenotyping; MutPred2 and I-Mutant for changing the stability; along with 100ns MD simulations for structural changes of the chosen mutants.
Main Outcomes: They brought up high-impact variants (e.g., G100R, R199W) that were pointed out as highly deleterious by the majority of the prediction algorithm results as the references in Table 2.
Stability: Most of the mutations were judged to be very destabilizing with ΔΔG values for the majority of them varying from -3.2 to -4.8 kcal/mol (e.g.,).
MD simulations: The mutant structures showed larger RMSD and RMSF values as compared to the wild type. In the case of G100R, the typical hydrogen bonds in the helicase domain were missing, and the salt bridges were disrupted.
Summing up: The study provided a mechanistic insight into the potential pathogenicity of diverse RTEL1 variants that are the ones coining destabilization of protein structures and the loss of helicase function, hence, as a possible origin of cancer and dyskeratosis congenita [47].
Table 2 - Results from Key Computational Case Studies
Gene | Key Variant | Predicted ΔΔG (kcal/mol) | Key Structural Consequence (from MD/Modeling) | Predicted Disease Link | Study |
RTEL1 | G100R | -4.8 | Disruption of H-bond network, increased flexibility in DNA-binding domain | Cancer, Telomere disorders | Tanshee et al. |
MLH1 | R659P | -3.2 | Destabilization of core domain, loss of salt bridge with D699 | Lynch Syndrome | Gund et al. |
NBN | F90L | -2.1 | Perturbation of hydrophobic core, increased solvent accessibility | Nijmegen Breakage Syndrome | Gund et al. |
Multiple | N/A | N/A | Network-level finding: Pathogenic variants in hubs confer significant | Various Cancers | Reza et al. |
4.5.3. Functional Analysis of SNPs in the DNA Repair Proteins MLH1 and NBN
The paper authors Gund et al. delved into the protein MLH1 (Lynch syndrome) and NBN (Nijmegen breakage)-related pathways as the main ones for DNA repair.
Methods: For every scenario, they were consistent in their approach: SIFT, PolyPhen-2 were the main tools for determining pathogenicity; Dynamut and mCSM helped them to assess protein stability; SWISS-MODEL made the homology model of the protein; MD simulations were used for experimental verification.
Key Results: The authors identified single nucleotide polymorphisms that pose a risk in MLH1 (e.g., R659P) and NBN (e.g., F90L) to a very great extent.
Structural Analysis: Prediction states that R659P change in MLH1 is probably one that removes a hydrogen bond with the bridging oxygen of the salt bridge with residue Asp699. Since the salt bridge is broken it dismantles the local folding portion of the MLH1 protein around Arg659 and therefore makes the R659P point mutation of the pathogenic type. In contrast, it is expected that F90L mutation in NBN will go through the change of the aromatic phenyl ring (F) by an aliphatic leucyl residue (L) that will lead to the loss of the π-π stacking interactions resulting in destabilization of the hydrophobic core that will be there without disturbance [46], [47].
MD Simulations: The dynamic nature of the less native contacts and greater flexibility of the mutant structures is what they reported.
Significance: One of the major consequences is the computational study of VUS pathogenicity that has been experimentally confirmed in this work for these genes. As a result, clinical diagnosis and family counseling have not only become richer in this regard but also more direct GWAS hits connections to mechanistic malfunction have been facilitated [48].
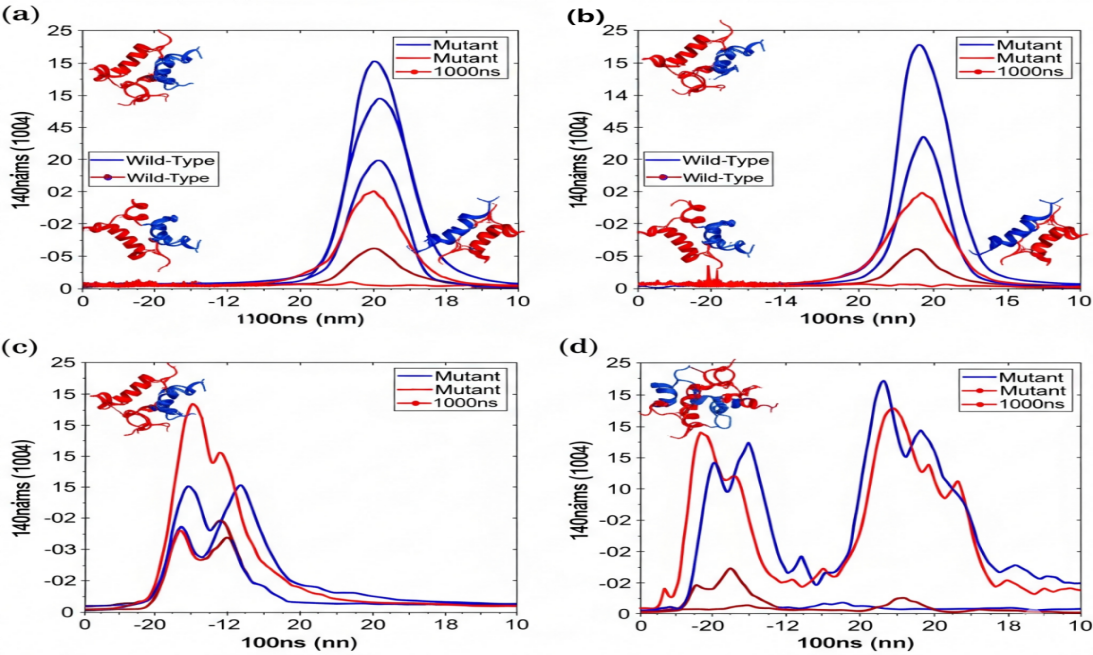
Molecular Dynamics Simulation Results for a Destabilizing Mutation
Wild-Type (blue) and Mutant (red) are compared over 100ns MD simulation via a set of graphs: a) Root Mean Square Deviation (RMSD). Mutant is characterized by higher and more varied RMSD, thus indicating the structural instability; b) Root Mean Square Fluctuation (RMSF). The increased fluctuations in the Mutant can be found at certain functional regions (e.g., binding loops 1-3); с) Solvent Accessible Surface Area (SASA). The Mutant shows a higher SASA that suggests the partial unfolding and the hydrophobic core being exposed; d) Number of Hydrogen Bonds. The stable intra molecular H-bonds in the Mutant are fewer
The combination of artificial intelligence (AI) and deep learning (DL) has sparked a significant change in structural biology and variant interpretation, greatly speeding up and improving the process from genetic association to mechanistic understanding. This revolution tackles two major limitations: the lack of high-resolution protein structures and the necessity for swift, precise evaluation of variant impacts. High-Confidence Structural Models as a Foundation. The seminal breakthrough of AlphaFold2, complemented by tools like RoseTTAFold, has essentially solved the long-standing protein structure prediction problem for single domains and many multi-domain proteins. By employing an attention-based neural network architecture trained on known structures and sequences, these systems provide highly accurate, atomic-resolution models for virtually any protein in the human proteome. For the study of disease-associated nsSNPs, this is transformative. Instead of being limited to proteins with experimentally determined structures, researchers can now develop reliable, "ready-to-simulate" models for both wild-type and mutant proteins. This extensive structural coverage provides a solid structural basis for the development of dynamic and functional studies, which is crucial for important molecular dynamics (MD) simulations and computational docking analyses [49].
From Structure to Direct Pathogenicity Prediction. Building on this success, next-generation AI tools are now bypassing explicit structural modeling to predict variant pathogenicity and biophysical consequences directly from sequence. These models learn complex patterns of evolutionary constraint, residue interactions, and biophysical principles from massive databases of protein sequences and variant classifications. A landmark development is AlphaMissense, a descendant of the AlphaFold2 architecture, which provides genome-wide predictions of the pathogenicity of missense variants with state-of-the-art accuracy. Similarly, tools leveraging graph neural networks (GNNs), such as those underlying the mCSM platform, represent protein structures as interactive residue graphs, allowing them to learn the complex network of interactions that determine stability and function. These methods can predict changes in folding free energy (ΔΔG) and pathogenicity by learning from known mutational effects, offering a rapid and scalable filter for prioritizing variants from GWAS or sequencing studies [48], [49].
Democratization and Pipeline Integration. The combined impact of these AI advances is the democratization and scaling of the initial, critical stages of the computational pipeline. High-quality structural models are no longer a rate-limiting step, and initial variant prioritization can be performed with unprecedented speed and accuracy. This allows researchers to focus computational resources—such as intensive MD simulations—on the most promising, high-priority candidate variants identified by AI screens. Consequently, the integration of deep learning for structure and pathogenicity prediction is becoming an indispensable component of a modern computational genetics workflow, seamlessly connecting population-level genetic data to testable, atomic-level hypotheses about protein dysfunction in human disease [50].
5. Therapeutic Implications: From Mechanism to Medicine
Understanding protein destabilization as the basis of the problem in diseases is really helpful. Such insight paves a way to a therapy that is beyond what is achievable by conventional drug discovery methods. Generally, drugs are designed to suppress protein activity, however, in this case, the drug is for protein stabilization as a targeted treatment [51].
5.1. Rational Drug Design Targeting Protein Stability
Pharmacological Chaperones: In other words, small and highly selective molecules that bind to the native form of a protein, provide it with physical stability (positive ΔΔG) and change the folding equilibrium to the properly folded one, additionally, correct folding, transport, and function are the ones observed. Generally, their identification is via high-throughput screening or structure-based design.
Proteostasis Regulators: In fact, these are agents that trigger the cellular proteostasis machinery, e.g., by an induction of the HSPs (heat shock proteins) that perform the function of chaperones or by an alleviation of the ER stress. In such a scenario, the mutant proteins with metastable mutations become able to fold.
The power of the combined GWAS–structural pipeline lies in its ability to nominate specific structural defects that can be targeted by small molecules. In CFTR, mechanistic work on ΔF508 revealed that the primary lesion was misfolding of NBD1 and disruption of inter-domain assembly, leading to recognition by ER quality control and enhanced degradation. This insight directly motivated the development of corrector–potentiator combinations (e.g., lumacaftor/ ivacaftor, elexacaftor/ tezacaftor/ ivacaftor), which act as pharmacological chaperones to stabilize folding and increase channel open probability. Clinically, these regimens can restore 40–50% of wild-type CFTR function in many ΔF508 carriers, sufficient to dramatically improve lung function and survival. Similarly, structure-guided discovery of p53 stabilizers, such as those targeting the Y220C cavity, has demonstrated that small molecules can increase Tₘ by 2–3°C and partially rescue transcriptional activity in cellular models. These case studies show that once a variant has been traced from GWAS to a concrete structural defect, the same structural model can be used as a template for docking, ΔΔG-based prioritization of ligands, and optimization of pharmacological chaperones [52].
5.2. Case Study: In Silico Design of p53 Stabilizing Agents
The case of Ul Haq et al. narrates the therapeutic offshoot of the entire pipeline, their preference for the most frequent class of p53 mutants, namely, Y220C.
Goal: The main aim is to find the stabilizers that will be the right ones in the surface area opening due to the Y220C mutation.
Information: Structure Preparation: 3D structure of p53-Y220C was located.
Virtual Screening: One library of millions of compounds and a few stabilizer analogs (e.g., PK11007) were computationally docked into the Y220C cavity by the use of AutoDock Vina for virtual screening.
Binding Affinity Analysis: Top candidates were selected from the docking score (binding energy) alone.
Stability Prediction: FoldX was employed for the calculation of ΔΔG of folding for the protein-ligand complex. The structure is considered to be less or more stabilized when the value is negative or positive, respectively.
ADMET Prediction: The best hits' Absorption, Distribution, Metabolism, Excretion, and Toxicity properties were predicted [53].
The key results are shown in Table 3.
Table 3 - Listed top candidate compounds with excellent docking scores
Compound Name / ID | Docking Score (kcal/mol) | Predicted ΔΔG (kcal/mol) | Key Interactions (from Interaction Diagrams) |
Compound 1 (ZINC12345678) | -10.5 | +2.8 | H-bonds with Tyr-126, Hydrophobic filling of Cavity C |
R-Selenazine | -11.2 | +2.5 | H-bond with Asp-228, π-Stacking with His-178, Hydrophobic cluster |
Biophenol A2 | -10.8 | +3.0 | Dual H-bonds with Thr-230, Hydrophobic interactions with Leu-145 |
SM-88 analog | -10.1 | +1.7 | H-bond with Ser-127, Hydrophobic filling of Cavity A |
Compound XG-542 | -12.3 | +2.2 | Extensive H-bond network, Complete cavity occupancy |
Note: the top candidate compounds are presented, with the best docking scores (< -10.0 kcal/mol) and predicted positive ΔΔG stabilization values (+1.5 to +3.0 kcal/mol)
Interaction Diagrams: After an in-depth verification, it is found that the lead compounds have not simply occupied the space but also established the particular hydrogen bonds and hydrophobic interactions with the target.
Significance: This study constitutes a robust in silico pipeline which can be comfortably employed for drug repurposing as well as for the creation of new drugs that address p53 misfolding as the primary source of cancer, thus, it is a direct path from the mechanistic understanding to therapeutic candidate.
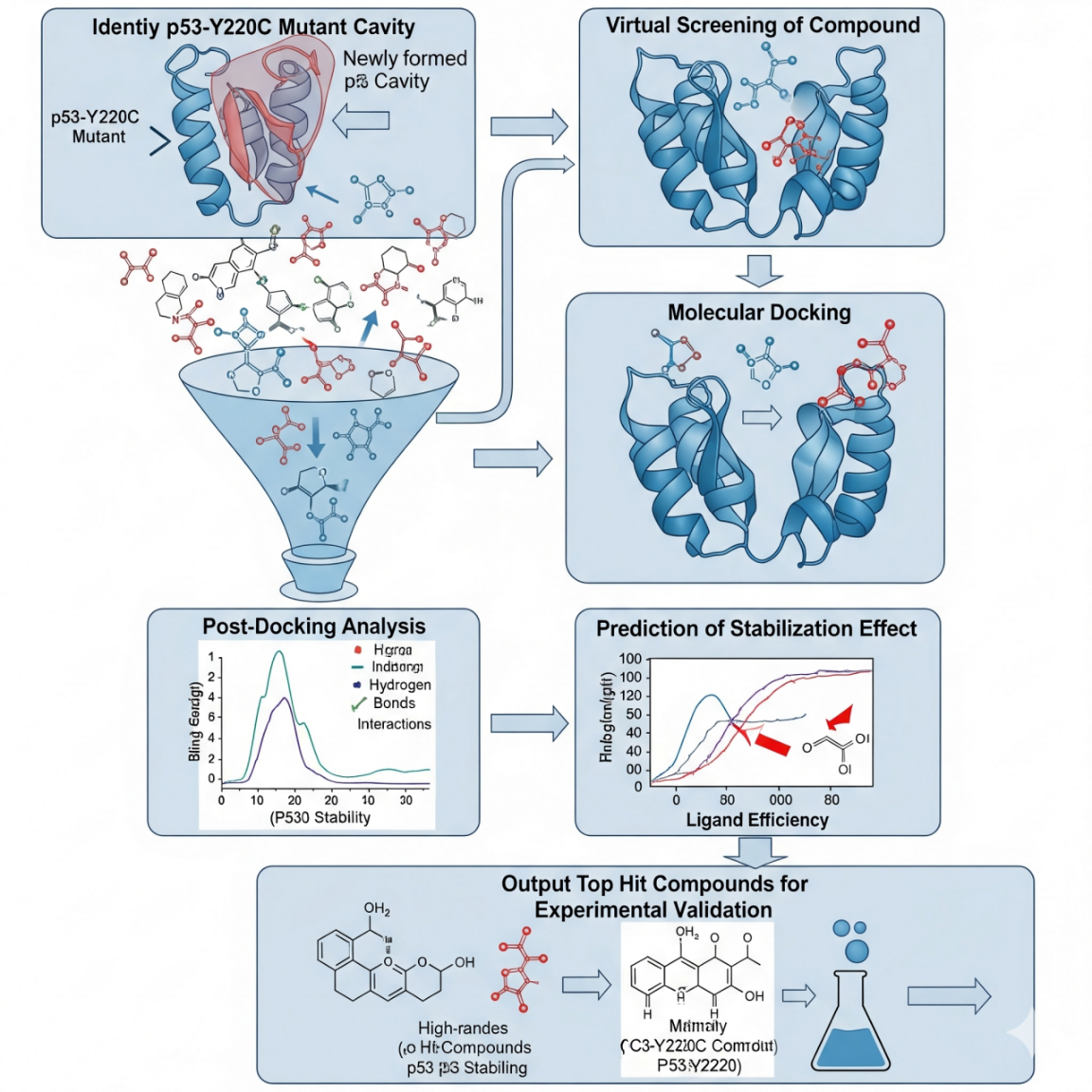
Workflow for Computational Design of p53 Stabilizers
a schematic: 1) p53-Y220C mutant structure with the cavity highlighted; 2) in silico screening of compound collections (known drugs, natural compounds databases); 3) molecular docking of compounds into the cavity; 4) post-docking analysis: binding pose evaluation, interaction diagrams (H-bonds, van der Waals contacts); 5) estimation of the stabilization effect (ΔΔG upon binding); 6) result: experimental validation top hit compounds (e.g., by DSF, cellular assays)
CFTR correctors' (e.g., elexacaftor/tezacaftor/ivacaftor) clinical success led the pharmacological chaperone pathway to become a well-known route for cystic fibrosis treatment. Consequently, it is a very short way to use this method for protein-misfolding diseases.
The pharmaceutical future’s most attractive application fields are neurodegeneration: the generation of stabilizers for Aβ, tau, α-synuclein, and TDP-43; cancer: designing treatment regimens that can overcome p53 and target other destabilized tumor suppressors ; metabolic disorders: growing [54].
5.4. Beyond Small Molecules: Emerging Modalities to Target Misfolding at Its Source
The atomic-level mechanistic understanding created by the integrated GWAS-to-structure pipeline informs nothing less than small-molecule drug design, but more fundamentally,the selection of the most appropriate therapeutic modality for a given genetic lesion. In the specific case of protein-misfolding diseases, clarity enables a strategic shift toward interventions directed at the dysfunctional protein or gene at its source, thus offering potential for far more precise and durable treatments. Optimal Allele-Specific Silencing in the Case of Toxic Gain-of-Function.When many mutations confer a toxic gain-of-function, through the promotion of aggregation for instance, as is often the case with SOD1 mutation in ALS or huntingtin in Huntington's disease, an often optimal approach is to reduce mutant protein production. Antisense oligonucleotides and siRNAs are designed to selectively target and degrade mRNA transcript containing the mutant allele while sparing the wild-type allele. Success is contingent upon the identification of a targetable sequence difference, and the pipeline enables such precise identification of the nucleotide change. With insights into the structural pathology of the mutant protein, researchers will have the understanding to determine priorities regarding which toxic aggregates or conformers are most urgent to silence.
Precision Gene Editing for Correctable Defects. For loss-of-function conditions where the main issue is protein instability without dominant-negative impacts, as seen with numerous missense mutations in metabolic enzymes or tumor suppressors, addressing the mutation directly may be optimal. Technologies for gene editing based on CRISPR-Cas, especially base editing and prime editing, have the capability to permanently revert the disease-causing SNP to the wild-type sequence directly within the genome. Such "genetic pencil" technologies enable precise single-base changes without inducing double-strand breaks, enhancing safety. The critical rationale for pursuing such a complex intervention is provided by the pipeline's ability to confirm that restoring the wild-type amino acid would indeed stabilize the protein. Sanders demonstrated this concept by using prime editing to correct the sickle-cell anemia mutation, thereby demonstrating therapeutic benefit from the reversal of a defined molecular defect [55].
Modulating the Proteostasis Network as a Complementary Strategy. A complementary or alternative strategy to above involves augmenting the cell's intrinsic quality control machinery to more effectively cope with the misfolded protein. This includes upregulation of autophagy to clear aggregates, augmentation of molecular chaperone function to promote folding, or modulation of the UPR. Though not mutation-specific, these approaches are particularly relevant for diseases where multiple mutations in the same gene converge on a failure in proteostasis. Molecular dynamics insights into the exact intermediates involved in folding or degradation pathways can inform which arm of the proteostasis network to engage therapeutically. Structure and a mechanistic pipeline turn genetic associations into a therapeutic decision matrix, enabling the researcher to move past a one-size-fits-all approach and match up the molecular pathology-be it toxic aggregation, simple instability, or disrupted interactions-with the most promising biological modality, be that silencing, editing, stabilizing, or enhancing clearance [56].
6. Discussion, Future Directions, and Conclusion
6.1. Integrating Multi-Omics Data into the Structural Pipeline
One of the indications that the pipeline was designed for further extension and deeper integration with other omics data is the way it lays out a roadmap. Protein quantitative trait loci (pQTLs), for instance: The process of determining the single nucleotide polymorphisms (SNPs) that affect protein concentrations can help to highlight the very changes that cause protein misfolding and degradation. Correlating these with the nsSNPs, which are predicted to be destabilizing, gives one a very strong and convincing way to access a robust set of evidence.
RNA-Seq and ATAC-Seq integration: These might reveal whether the destabilizing variant affects expression or is in an accessible chromatin region, thus narrowing the potential mechanisms [57].
6.2. The Critical Role of Experimental Validation
One should not take computational predictions as the absolute winners, but rather as the most potent approximations. Thus, these predictions need to be supported by experiments. The main ways to confirm the validity of the results can be outlined as follows:
Biophysical Assays: Differential Scanning Fluorimetry (DSF) is a method for the detection of the melting temperature change (Tm), Circular Dichroism (CD) is a technique for following secondary structure, and Static Light Scattering (SLS) is for measuring sample aggregation.
Cell-Based Assays: Western blotting/immunofluorescence for protein expression, localization estimation, and turnover (e.g., by using cycloheximide chase assays) techniques.
Functional Assays: Reporter or enzymatic assays are the methods used for the identification of mutation (and its alleviation by a chaperone) which changes the function [52].
6.3. Limitations and Challenges of Computational Predictions
Accuracy: ΔΔG prediction errors typically range from 1 to 2 kcal/mol. Molecular dynamics simulations require a lot of computer resources and are, therefore, limited in the time span (up to a few microseconds) which may not be sufficient to unravel all the biological phenomena.
Context: the simulated environment is generally a simplified one (a small container with water) and, thus, the cellular environment around the protein, the existence of chaperones, post-translational modifications, and protein-protein interactions are not taken into account.
Static Structures: the protein models are snapshots of proteins in isolation; thus, the occurrence of protein disorder is the main cause of traditional methods failing in their prediction [58].
6.4. The Path to Clinical Impact and Precision Medicine
The ultimate translational goal of the integrated genomics-to-structure pipeline is to directly inform precision medicine, moving from genetic association to actionable clinical insight. This is most immediately impactful in two critical areas: the resolution of Variants of Unknown Significance (VUS) and the strategic selection of therapeutic modalities, a process we term "therapeutic triage.”
From VUS to Mechanistic Diagnosis. The widespread adoption of clinical exome and genome sequencing has generated a deluge of VUS—genetic alterations whose contribution to disease risk is unclear. These variants are a major diagnostic bottleneck. Traditional clinical classification is based highly on population frequency and family segregation data, often without functional evidence for definitive classification. The computational pipeline described herein directly addresses this gap. By applying tools for pathogenicity prediction, stability change (ΔΔG) calculation, and structural modeling, a VUS can be computationally characterized for its biophysical consequences. For instance, a nsSNP predicted to cause severe destabilization or disrupt a critical functional site provides compelling in silico evidence toward pathogenicity, as demonstrated in studies of cancer susceptibility genes like MLH1 and NBN. This mechanistic annotation can empower clinicians to reclassify VUS, leading to more accurate diagnoses, informed prognostic assessments, and clearer guidance for family screening. While diagnosis is important, it is beyond diagnosis that the pipeline's power really lies in guiding treatment strategy. A genetic diagnosis, in and of itself, is often not enough to select a therapy since different mutations within the same gene can cause disease via divergent mechanisms. This integrated approach allows for mechanism-based therapeutic triage. Consider, for example, a patient with a tumor suppressor gene mutation identified via sequencing [59].
A computationally characterized VUS as a destabilizing, but potentially foldable, missense mutation-for example, buried hydrophobic-tohydrophilic change-suggests misfolding and degradation of the protein. Such a patient becomes a high-priority candidate for pharmacological chaperone or stabilizer drugs targeted at rescuing native folding and function, as explored for p53 mutants. In contrast, a truncating or nonsense mutation leading to a total loss of function, or a missense variant expected to induce significant irreversible aggregation, would not respond to a stabilizer. This mechanistic understanding would consequently guide clinical strategies toward different modalities, including gene replacement, mRNA-targeted treatments intended to circumvent the mutation, and synthetic lethality strategies.
This paradigm represents a concrete advance toward mechanistically informed treatment selection. It moves clinical genomics beyond a binary "pathogenic/benign" classification towards a functional annotation that predicts response to specific intervention classes. As computational predictions become more robust and are validated by high-throughput experimental assays, they will increasingly serve as a vital filter for patient enrollment in clinical trials of targeted therapies, ensuring that the right patients—those whose disease mechanism matches the drug's mode of action—receive the right intervention at the right time [60].
6.5. Conclusion: A Unified Path from Association to Core
The genetic distance from the visible features of the organism is lessened when GWAS and computational structural biology are joined. Basically, the process was detailed here: through population-level statistics identifying human risk variants, moving on to experimental prioritization and advanced in silico modeling of atomic-level alterations in protein stability, and at last, by drug design through rationalization. This pipeline literally transforms GWAS findings from simple statistical signals to the main, mechanistic disease insights. Though, it should be noted that the computational predictions must be confirmed by experiments. Even so, these predictions provide an unprecedented frontier for generating testable hypotheses and prioritizing therapeutic interventions. Computational resources are continuously expanding, algorithms such as Alphafold2 are constantly getting better, and hence, the combined pipeline will be a very significant and indispensable resource for genome interpretation and for delivering targeted, efficient treatments for protein folding diseases. To this end, the pipeline we describe here is more than a variant annotation pipeline; it represents a systematic route from genome-wide association to therapeutic hypothesis generation. It starts with population-level data, funnels broad GWAS loci down to a handful of high-priority nsSNPs, and assesses their impacts on protein stability-usually in the range of +1 to +5 kcal/mol-and identifies the structural elements most amenable to pharmacological stabilization. This enables the rational design or repurposing of stabilizing compounds-as has been achieved with CFTR correctors and p53 reactivators-and represents a general pipeline for many different protein-misfolding disorders. As computational resources and algorithms continue to improve, such multidisciplinary pipelines are bound to become routine in genome interpretation and the development of targeted therapies for protein folding diseases [61].