ALIGNMENT OF PSEUDOREADS OBTAINED FROM HOMOLOGOUS SEQUENCES IN IDENTIFYING POTENTIALLY TOLERATED GENOMIC VARIANTS
ALIGNMENT OF PSEUDOREADS OBTAINED FROM HOMOLOGOUS SEQUENCES IN IDENTIFYING POTENTIALLY TOLERATED GENOMIC VARIANTS

Abstract
The use of Next-Generation Sequencing (NGS) has proven to be clinically beneficial, but it has also revealed a significant number of variants that we are unable to accurately define and categorize in terms of pathogenicity. These variants are known as variants of uncertain significance (VUS) which are detected en masse in each NGS run. Unlike amino acid substitutions and splice site mutations, common variants in non-coding regions have not been extensively studied and are still mostly classified as VUS. In this paper, a new concept was proposed to identify potentially tolerated variants, including variants in non-coding regions, based on the Genetic Alignment of “Pseudoreads” from Homologs (GAPH) method. We have discovered a total of 5,859,205 variants, the majority of which have never been documented in the largest population database, GnomAD, and only 0.0015% (88 variants) were classified as pathogenic according to the ClinVar database. Overall, the results of this study demonstrate the efficacy of our new method to refine a variant tolerability, many aspects of which could be further adjusted to optimize the results.
1. Introduction
Molecular diagnostics has undergone a stage of rapid development and growth in the last decade, intensively increasing the amount of genomic data , . However, the clinical significance of the majority of variants being detected is unknown. Variants in non-coding regions, which account for about 98% of the genome, are mostly classified as variants of uncertain significance (VUS) . There is increasing evidence that non-coding variants in different diseases, including cancer, are becoming more recognized as contributors to the development of these diseases , , , .
Computational variant effect predictors leverage the vast amount of biological data currently available to infer the fitness effects of human variants. The American College of Medical Genetics and Genomics (ACMG) previously developed the guidance for interpreting sequence variants, which involves utilizing in silico predictors to classify neutral (benign, BP4 criterion) and pathogenic (pathogenic, PP3 criterion) variants .
The majority of widely used and well-established computational predictors were designed to analyze protein sequences or splicing sites and, therefore, cannot be applied to variants in non-coding regions. Nevertheless, there are multiple tools that predict the deleteriousness of non-coding region variants based on the evolutionary conservation of studied genomic fragments , , , . Other variant effect predictors, which can analyze variants in non-coding regions, are based on the annotation data , , . However, none of these existing tools are capable of taking into account the specific sequence changes observed during evolution.
For such a reason, we have developed a new method, Genetic Alignment of Pseudoreads from Homologs (GAPH), that identifies differences between human genes and genomic sequences of various species. We have also evaluated whether this approach is appropriate and relevant enough to use it for variant tolerance prediction.
2. Research methods and principles
263 cancer-related genes were obtained from the COSMIC database , and the longest protein isoform was derived for each gene from the NCBI RefSeq database . These amino acid sequences were then queried against the RefSeq protein database using BLAST with an E-value threshold of 10 and a maximum number of resulting sequences set to 1000, and the corresponding nucleotide sequences were obtained.
The resulting gene sequences were fragmented into 70 nucleotides pseudoreads with a 35-nucleotide overlap. Each nucleotide was assigned a Phred-score of 30. The resulting pseudoreads were aligned to the originally queried human gene sequence using BWA-MEM with default parameters: according to the original BWA paper, only reads with no more than 4 mismatches or gaps were mapped to the reference sequence . Finally, variants were called using VarScan v2.4.4 with at least two reads support and a minimum of eight read-depth coverage required.
GAPH-resulted variant set was compared with known benign and pathogenic variants from ClinVar (accessed on July 14, 2022) , as well as variants from GnomAD v3.1.2 (accessed on June 3, 2023) .
Since benign variants tend to have a relatively high population allele frequency, we have used a subset of the GnomAD v3.1.2 database along with its full version, which included variants with population allele frequency > 0.01% only.
3. Main results
Using GAPH, we identified 5,859,205 nucleotide variants that are present in homologous sequences and therefore might be evolutionarily permissible. Of the 10,368,623 variants of the studied genes found in the GnomAD v3.1.2 database, 965,593 (9.3%) were in the resulting set. A subset of the GnomAD database was created, consisting of 569,998 variants with a minor allele frequency (MAF) greater than 0.1%, which accounted for 5.5% of all variants in GnomAD. Out of these, 72,778 variants were found in the GAPH-resulted set, making up 7.5% of GnomAD variants with MAF > 0.1% and 1.2% of the entire GAPH variant set (figure 1).
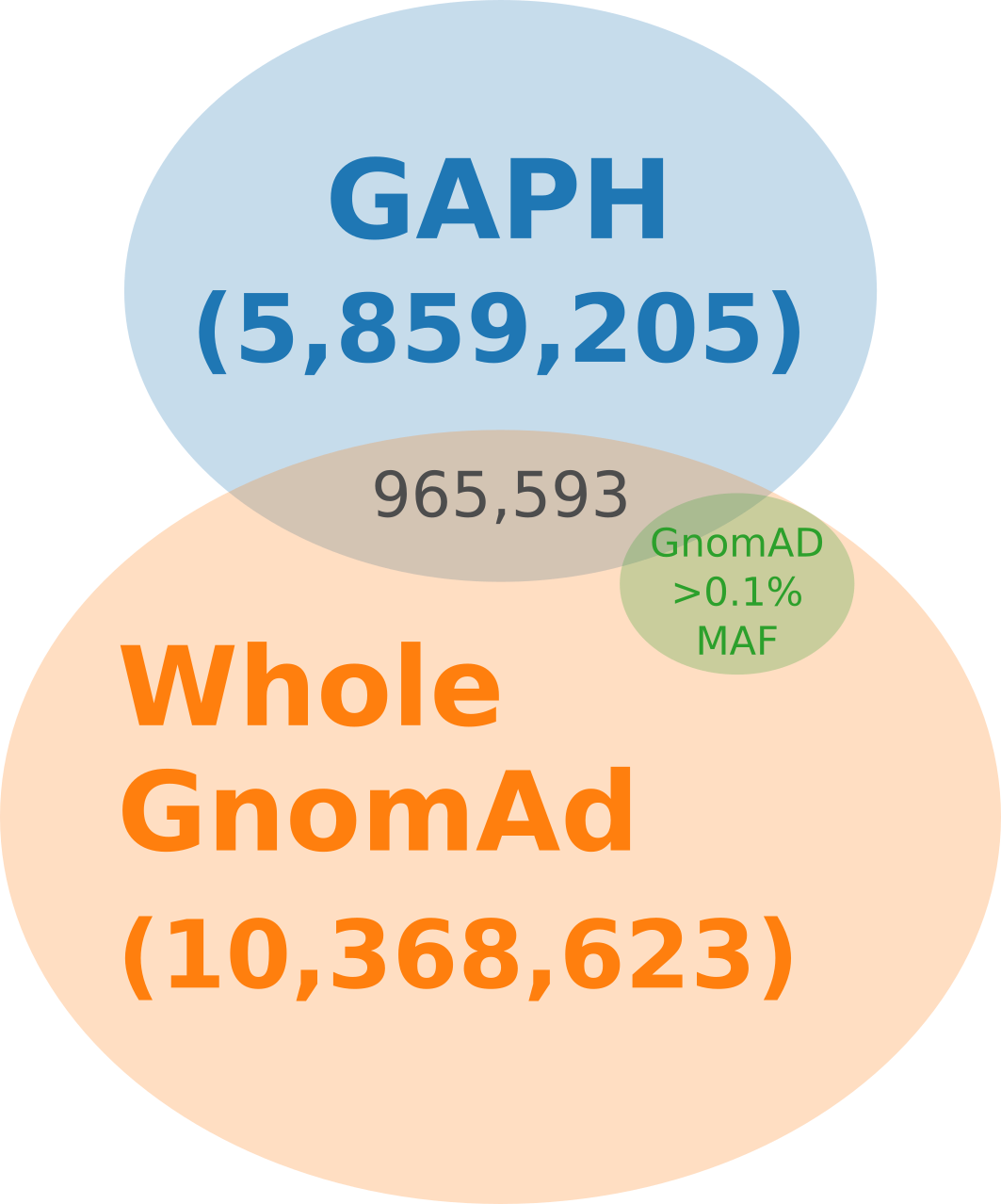
Figure 1 - Venn diagram illustrating the overlap between GAPH-resulted set, variants found in GnomAD v3.1.2, and subset of GnomAD variants with minor allele frequency (MAF) > 0.1%.
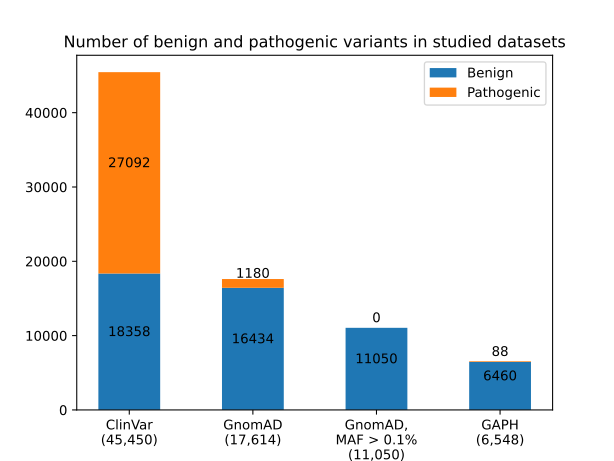
Figure 2 - Distribution of benign and pathogenic variants from ClinVar, GnomAD v3.1.2, GnomAD subset with MAF of variants > 0.1%, and the GAPH-resulted variant set
Note: the total number of benign and pathogenic variants is indicated in parentheses
4. Discussion
In this paper, we have demonstrated GAPH, a method for obtaining a set of variants that are present in homologous sequences from a nucleotide sequence alignment. The core element of this approach is a pseudoreads usage, which is a concept previously applied in transcriptomic studies , and genome assembly software . To our knowledge, this is the first study utilizing pseudoreads for variant effect prediction, and that is why this report is focused on presentation of the method concept and its general applicability with no intention to conduct an exhaustive study. Many additional aspects of it might be adjusted further to achieve a desirable level of accuracy, combined with validation and verifications studies.
We used amino acid sequences as queries for the search of homologous sequences, which is a common approach , . Despite our relatively high E-value threshold of 10, we further implemented the BWA-MEM algorithm, which prevents mapping of 70 nucleotide-long pseudoreads with more than 4 mismatches. However, our method, as well as the current variant effect prediction tools, does not take into account orthologous/paralogous relationships, which reduces the overall performance of categorizing disease-causing and benign variants .
For example, a comprehensive phylogenetic study of the BRCA2, TP53, DICER1, PIK3CA, and BRCA1 genes decreased the number of variants obtained through GAPH in these genes from 77,154 to 72,505. It also reduced the number of matches with GnomAD from 12,345 to 11,484, which might aid to decrease the total number of variants erroneously classified as potentially tolerated (unpublished). The precise identification of orthologs for major disease-causing genes might be further introduced in our method.
There are also several widely used nucleotide sequence alignment tools, e.g. Bowtie and BWA-SW . While BWA-MEM is the most modern algorithm in the BWA package, which is utilized by popular biological pipelines like GATK and SPAdes , other alignment tools exist that could potentially enhance the performance of the method presented here. The operational length of a pseudoread was chosen based on the minimum allowable read length for alignment using BWA-MEM but it also might be adjusted for the better performance.
The main flaw of this method is that identification of damaging variants is not available, similarly to the GnomAD population database . Therefore, our approach is intended to be used similarly to GnomAD in order to identify only potentially tolerated variants. However, there is a significant difference between the GnomAD database and GAPH-resulted variant set: only less than 10% of variants are present in both sets (figure 1). We propose that such a discrepancy is likely due to the disparity in the genomic diversity data used from various sources: while GnomAD is based on the study of the human genome variation, GAPH specifically focuses on sequences of homologous genes in various organisms.
We understand that not all GnomAD variants should be treated as benign, and there is a discrepancy regarding the population allele frequency threshold that should be applied to minimize the probability of a variant pathogenicity . We used a threshold of 0.1% MAF to evaluate the predictive capability of this GnomAD subset and compare it to our dataset.
Overall, the current paper demonstrates the potential of our novel method to generate a database of potentially tolerated variants, which utilizes phylogenetic information unlike all human population databases.
5. Conclusion
This paper presents a concept of new methodology for obtaining a set of potentially tolerated variants from an alignment of pseudoreads generated from sequences of homologous genes. Our proposed approach has enabled us to identify numerous new variants in the homologs of the genes under investigation. These variants can be used in a similar manner to those found in population databases such as GnomAD. However, while GnomAD relies on genetic variation only in human genes, our method utilizes sequences of homologous genes in different organisms. Since GAPH-resulted set is based on a different principle, it can be used together with GnomAD for variant tolerance prediction.
We are still at the early stages of exploring GAPH, and the potential for adjusting various aspects to enhance its performance is under ongoing study. For example, a comprehensive deep phylogenetic analysis could be applied additionally, a different alignment algorithm could be tested, and the pseudoread length could be further modified to improve performance and applicability.