Машинное обучение с применением множественной логистической регрессии для предсказания антимикробных и гемолитических пептидов и их обнаружения в крупных белках
Машинное обучение с применением множественной логистической регрессии для предсказания антимикробных и гемолитических пептидов и их обнаружения в крупных белках

Аннотация
Антимикробные пептиды (АМП) рассматриваются в качестве перспективного пула альтернативных антимикробных агентов в пост-антибиотическую эру. Поскольку ряд ограничений, в особенности цитотоксичность, лимитирует их имплементацию в клинику, поиск новых нетоксичных АМП имеет высокую актуальность. В настоящем исследовании мы применили множественную логистическую регрессию для предсказания антимикробной и гемолитической способности пептидов. Две построенные модели продемонстрировали приемлемую предсказательную силу (аккуратность, чувствительность, специфичность, точность, F-мера ≥ 0,82, ROC AUC > 0,91 при оцененных оптимальных пороговых уровнях). Модель для предсказания антимикробной активности была далее применена для обнаружения возможных АМП в последовательностях крупных белков. Валидация метода была осуществлена на предшественниках известных АМП различных структурных классов – нейтрофильный пептид человека 1 (HNP1), кателицидин LL-37, а также тахиплезин I. Во всех случаях локализация зрелых АМП была предсказана верно, т.е. на C-конце (HNP1, LL-37) или в середине последовательности предшественника (тахиплезин I). Исследование обеспечивает простой для интерпретации метод предсказания антимикробной и гемолитической активности пептидов и их идентификации в крупных белках.
1. Introduction
The antibiotic resistance crisis is considered as a major challenge for modern medicine and raises questions of obtaining alternative antimicrobials , . Antimicrobial peptides (AMPs) represent a group of promising candidates due to their high anti-pathogen activity in the low micromolar range of concentrations, a broad spectrum of targets and a low potential to provoke bacterial resistance , . AMPs act as effector molecules of innate immunity of virtually all living organisms. Although AMPs are a highly diverse and difficult-for-classification group of biologically active peptides, most of them possess a common set of physicochemical properties. Typically, they are amphipathic peptides with spaced clusters of cationic and hydrophobic amino acid residues (a.a.). The primary mechanism of their action includes adsorption on negatively charged molecules of pathogenic surface due to electrostatic interactions and insertion into the lipid bilayer of the cell. The pore formation results in efflux of vital ions and metabolites and eventually to the target cell death, although alternative mechanisms of AMPs action have been described , .
The main and well-described AMPs of human innate immunity are cathelicidin LL-37 and human neutrophil peptides (HNPs) .
In spite of indisputable advantages, AMPs have not yet become a commonly prescribed antimicrobial drugs. One of the recognized limitations of AMPs implementation is cytotoxicity, i.e. the action on host cell membranes. In most pipelines of AMPs elaboration, cytotoxicity is evaluated in hemolytic assays . Clinically prospective AMPs are expected to have high selectivity of action.
In the context of the AMPs action as a double-edged sword, databases of antimicrobial and hemolytic peptides (HPs) have been created. The databases and existing predictive tools are reviewed in , , ; a more detailed review passage on tools for cytotoxic peptides' prediction can be found in a recent research paper . Experimentally validated peptides’ antimicrobial and hemolytic action is a tempting pool of data for machine learning (ML). Methods of AMPs prediction using ML include support-vector machine, hidden Markov models, random forest, artificial neuronal networks, decision tree, K-nearest neighbor, linear discriminant analysis, in addition to others. While quantitative models intend to predict minimum inhibitory concentration (MIC) or analogous numeric characteristics of a peptide’s activity, qualitative models are binary-type tools distinguishing between AMPs and non-AMPs or HPs and non-HPs. The qualitative approach seems to have advantages since protocols of antimicrobial and hemolytic activity measurement are not strictly standardized, and exact active concentrations can vary greatly depending on experimental conditions, a specific target, i.e. microbial strain or a source of red blood cells, etc. Logistic regression (LR) is not a widely spread method of prediction among available servers, but it is easy for use and interpretation and has been used for AMPs prediction .
Search of novel AMPs may build on known sequences of large proteins. Some of them are precursors of well-described AMPs typically containing a signal peptide at the N-terminus and a fragment corresponding to a mature AMP (in addition, other domains can be present). Nevertheless, peptides with antimicrobial activity may be derived from large proteins without described direct antimicrobial action. This may occur under physiological proteolysis, as it was demonstrated for anaphylatoxins, i.e. the derivatives of complement proteins C3, C4, C5 , , , , and α-melanocyte stimulating hormone, i.e. the proopiomelanocortin derivative . Identification of AMPs in large proteins utilizes the concept of a sliding window to screen the whole input sequence. Such type of peptides search is available via some web-servers. However, they have some disadvantages. The AMPA server is adapted for narrow sliding windows and, in our experience, does not predict the location of actual active fragments even in such classic AMPs as human defensins and cathelicidin LL-37-related peptides. Another server, Antifp is restricted to the prediction of antifungal peptides. Moreover, to our knowledge, LR has never been applied for revealing promising fragments in proteins sequences.
Since we conclude that there is a lack of easy-for-interpretation and validated for a broad spectrum of peptides models for AMPs and HPs prediction, including that from long sequences, we aimed to elaborate a new approach based on LR.
1.1. Abbreviations
a.a. – Amino acid residue; AAC – mino-acid composition; AMP – antimicrobial peptide; AUC – area under curve; df – degrees of freedom; HP – hemolytic peptide; LR – logistic regression; MIC – minimum inhibitory concentration; MCC – Matthews correlation coefficient; ML – machine learning; ROC – receiver operating characteristic; VIF – variance inflation factor
2. Research methods and principles
2.1. The general algorithm of the study
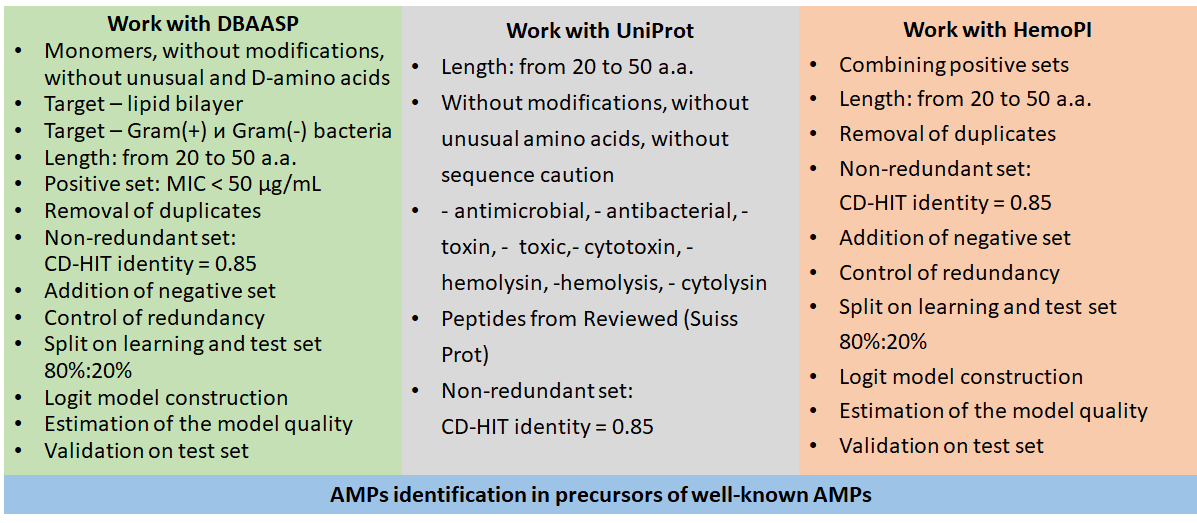
The general algorithm of the study
2.2.1. Antimicrobial activity: positive dataset
DBAASP (the Database of Antimicrobial Activity and Structure of Peptides) was used as a source of positive dataset containing naturally occurring and artificial AMPs . The database was filtered as follows: only monomeric peptides without modifications, unusual and D-amino acids, without “X” as an a.a., having MIC as described target activity value and MIC < 50 μg/mL, acting on lipid bilayer of gram-positive and/or gram-negative bacteria. The length from 20 to 50 was considered optimal. Too short and too long peptides were avoided for some reason. For example, amino-acid composition (AAC) value was intended to be used in ML (see Section 2.2.4) but a single a.a. in a very short peptide would give a very high AAC value and, at the same time, too many a.a. would have AAC value equal to 0. On the other hand, too long sequences are not prospective for synthesis and implementation. Besides, the use of sequences in a very wide window of lengths would make the dataset less uniform. Among duplicates (i.e., rows corresponding to one peptide with numerous target species) only one representative was retained while other representatives were excluded. Importantly, they were removed after MIC filtration was performed, so all active peptides were preserved in the positive set.
The data redundancy problem was solved by using CD-HIT, which is a widely used algorithm for sequences clustering and comparison , , . Instead of a standard program, the analogous cdhit() function from the CellaRepertorium R package was used. Before that, sequences were transformed to the AAString object format using the Biostrings package. Identity level 0.85 was used to generate a non-redundant dataset.
2.2.2. Hemolytic activity: positive dataset
The positive set of HPs was the assembled by combining positive datasets from the HAPPENN database : HemoPI-1 main positive, HemoPI-1 validation positive, HemoPI-2 main positive, HemoPI-2 validation positive, HemoPI-3 main positive, HemoPI-3 validation positive. Peptides of the length 20-50 were retained. CH-HIT was used to remove similar peptides (identity 0.85).
2.2.3. Negative dataset
The negative dataset was extracted from UniProt, which is a comprehensive database containing polypeptide sequences with functional information . Similarly to the positive dataset of AMPs, only 20-50-a.a peptides without modifications, unusual a.a., sequence caution were selected. Entries with keywords “antimicrobial”, “antibacterial”, “toxin”, “toxic”, “cytotoxin”, “hemolysin”, “hemolysis”, “cytolysin” were excluded. Then the reviewed database (SwissProt) was used for the final negative set formation. Duplicates and redundant peptides were excluded as described above. The negative dataset was used in the both antimicrobial and hemolytic ML models.
2.2.4. Performance of machine learning
In the both models, sizes of positive and negative datasets were equal, i.e. the datasets were balanced. This was reached by random exclusion of sequences from the negative dataset. For the both ML pipelines, CD-HIT was used to control possible similarities between positive and negative sets to ensure that they were different enough with the identity cut-off 0.85. Before the performance of ML, all datasets were randomized. All datasets were split to obtain a training set (80%) and test set (20%).
For all peptides, a number of properties was calculated. Peptides R package was used to calculate net charge at pH 7.0, hydrophobicity (scale Fasman was selected in preliminary models as leading to the best model performance), hydrophobic moment at the angle ϑ = 96° . Amino-acid composition (AAC) was calculated using protr package.
LR was used to estimate probability of antimicrobial or hemolytic capacity. LR is a statistical method applying sigmoid function operating with probabilities to linearize the data. Logit function of probability of a positive outcome
Therefore,
The optimal cut-off can be used to predict the outcome in a binary manner.
Features selection was a critical step in the modelling. It can be said that no rigorous algorithms were used to make a final list of predictors. However, several rules were used to construct models of acceptable quality. Since LR belongs to the class of generalized linear models, it is sensitive to multicollinearity. Correlated features were excluded in such a way to preserve as more as possible features. Methionine residues were excluded from the final list of the features since methionine represents the first a.a. in immature peptides from the negative sets downloaded from UniProt; inclusion of methionine residues would produce bias towards prediction of polypeptides with preserved signal peptides regardless their actual biological activity. ML using LR was performed using R function glm(). The most significant predictors were selected. After features selection, variance inflation factor (VIF) was calculated to finally make sure that no relations are present in the list of predictors. In the both models, the number of predictors was considerably less than the number of observations in both positive and negative groups. Besides, the models' fitness was estimated by χ2:
and p-value was calculated for degrees of freedom (df) equal to the difference between df in the null model (intercept-only model) and the model with predictors (in other words, to the number of predictors). Density plots were built to visualize the distribution of predictors between the positive and the negative group in training sets.
As the output of each of the models, formulae for calculation of LR probabilities (i.e., pAntimicrobial or pHemolytic) were obtained.
One way or another, predictive power rather than formal correctness was the main criterion of the models' quality. Models validation was performed on test sets, and standard parameters were calculated for estimation of the models quality using confusion matrices (TP – true positive, TN – true negative, FP – false positive, FN – false negative; MCC – Matthews correlation coefficient):
Interpretation of these parameters can be found elsewhere. The parameters were calculated for cut-offs 0.35, 0.4, …, 0.85.
ROC (receiver operating characteristic) analysis was performed using pROC R package. Density plots were built to visualize distribution of predicted pAntimicrobial or pHemolytic values by actual positive and negative groups.
2.3. Prediction of AMPs from precursor proteins
The model for AMPs prediction was applied for recognition of well-known AMPs in their precursor proteins. Mature peptides cathelicidin LL-37 (UniProt ID 49913), α-defensin HNP1 (UniProt ID P59665) comprise C-terminal parts of their precursor molecules. In contrast, tachyplesin I (UniProt ID P14213) is located in the middle part of its unprocessed precursor. The three peptides belong to different structural classes: LL-37 is α-helical, HNP1 contains 3 antiparallel β-strands, a disordered linker and is stabilized by three disulfide bonds; tachyplesin I is a relatively short β-hairpin. They also belong to different taxa – human and invertebrates, namely horseshoe crabs .
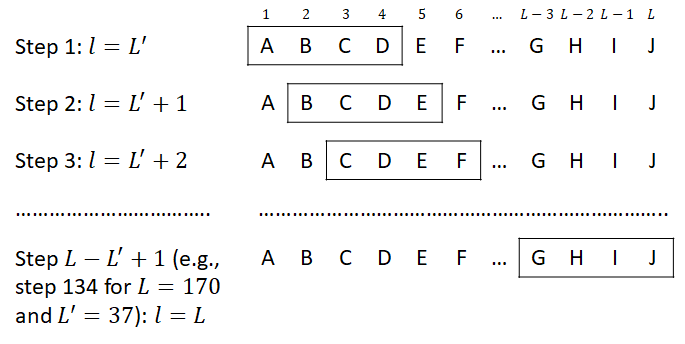
The sliding window applied to the AMPs recognition in proteins
L is the length of the peptide, L' is the length of the sliding window, l is the position of the C-terminal a.a. within the window
The study was performed using R language (v4.3.0) in the RStudio integrated development environment (2023.03.0) (R Core Team (2023)) . The following R packages were used: Biostrings v2.68.1, car v3.1-2 , CellaRepertorium v1.10.0, dplyr v1.1.2 , ggplot2 v3.4.2 , ggpubr v0.6.0 , Peptides v2.4.5 , pROC v1.18.2 , protr v1.6-3 .
3. Main results
3.1. Results of machine learning
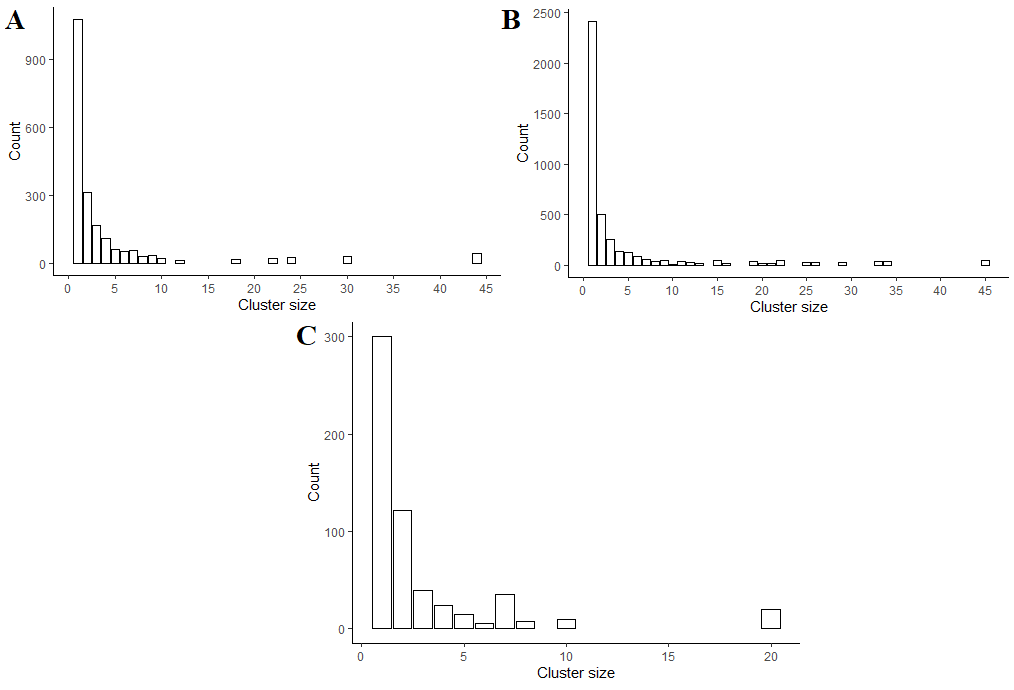
Distribution of cluster sizes in redundant datasets:
A – Antimicrobial positive; B – Negative; C – Hemolytic positive
Table 1 - Sizes of sets used in ML for antimicrobial activity prediction
Set | Split | Number of peptides |
AMP (1335) | Training set: 80% | 1068 |
Test set: 20% | 267 | |
Non-AMP (1335) | Training set: 80% | 1068 |
Test set: 20% | 267 | |
| Training set: 80% (2136) Test set: 20% (534) | Sum: 2670 |
Table 2 - Sizes of sets used in ML for hemolytic activity prediction
Set | Split | Number of peptides |
Hemolytic (390) | Training set: 80% | 312 |
Test set: 20% | 78 | |
Non-hemolytic (390) | Training set: 80% | 312 |
Test set: 20% | 78 | |
| Training set: 80% (624) Test set: 20% (156) | Sum: 780 |
Predictors used in the two models
green and red cells correspond to predictors with positive or negative effect on the dependent variable, respectively; AAC(X) – Amino-acid composition value of a residue X (name of an amino acid in standard one-letter code); n.s. – non-significant
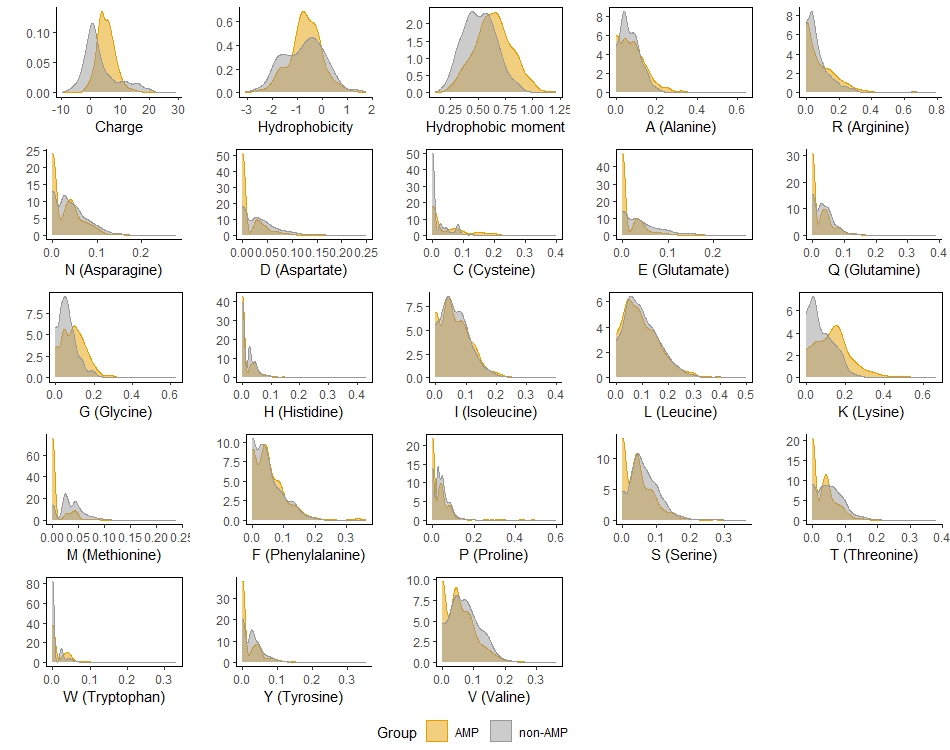
Distribution of all predictors (features) by the groups of AMPs and non-AMPs in the training set
plots of the selected predictors are bordered
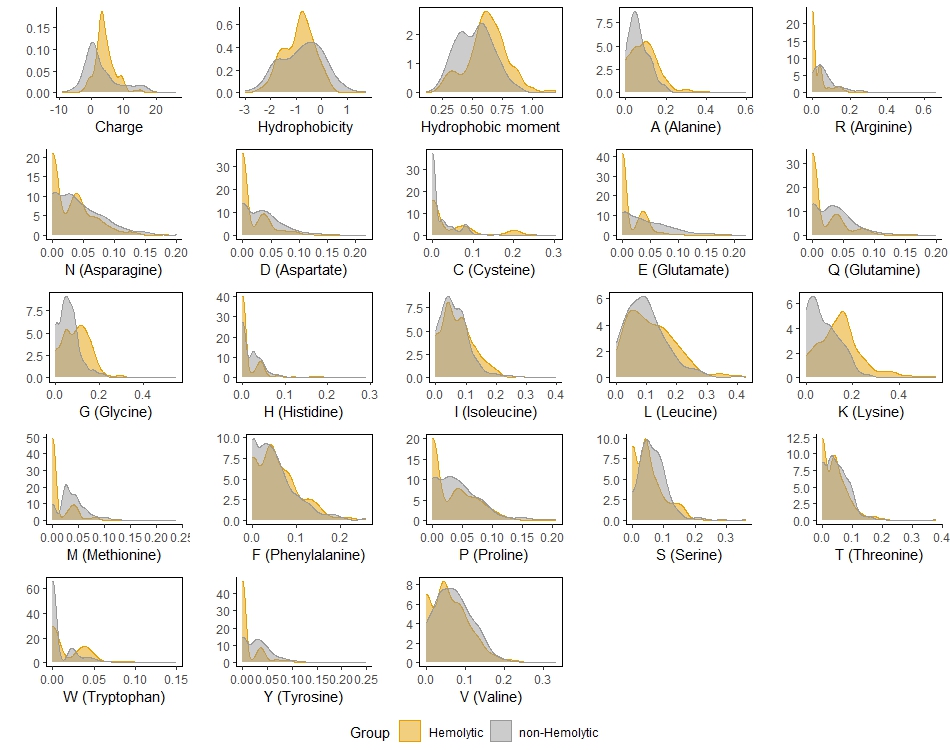
Distribution of the all predictors (features) by the groups of hemolytic and non-peptides in the training set
plots of the selected predictors are bordered
Table 4 - Essential parameters of the models
Parameter | Model | |
Antimicrobial activity prediction | Hemolytic activity prediction | |
Null deviance (null model) | 2961.1 | 865.05 |
df (null model) | 2135 | 623 |
Residual deviance (model with predictors) | 1610.6 | 418.09 |
df (model with predictors) | 2122 | 610 |
χ2 | 1350.5 | 449.96 |
df (number of predictors, i.e. df in the model with predictors – df in the null model) | 13 | 13 |
p-value | << 0.001 | << 0.001 |
Validation on test sets was performed. The estimated optimal (maximum accuracy) probability cut-off for antimicrobial activity prediction was 0.6; for hemolytic activity – 0.55. For antimicrobial activity prediction, accuracy = 0.86, sensitivity = 0.82, specificity = 0.90, precision = 0.91, F-measure = 0.86, MCC = 0.72. For hemolytic activity prediction, accuracy = 0.88, sensitivity = 0.87, specificity = 0.91, precision = 0.91, F-measure = 0.89, MCC = 0.77. Confusion matrices and the calculated parameters for cut-offs 0.35, 0.4, 0.45, …, 0.85 are represented in supplementary file Quality_of_prediction.xlsx (Sheets “Antimicrobial” and “Hemolytic”), and optimal cut-offs are colored in yellow for each of the models. ROC analysis was performed for estimation of the predictive quality of the two models (Fig. 6). ROC AUC for the first model was 0.914, and for the second model – 0.933. Briefly, the both models can be considered as adequately describing both antimicrobial and hemolytic activity.
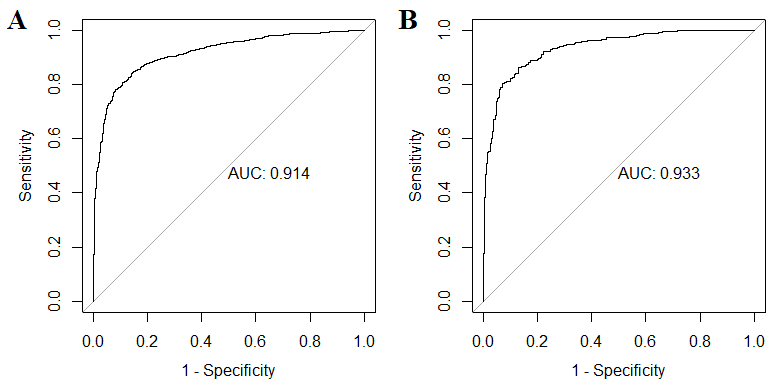
Results of ROC analysis of antimicrobial (A) and hemolytic (B) activity prediction
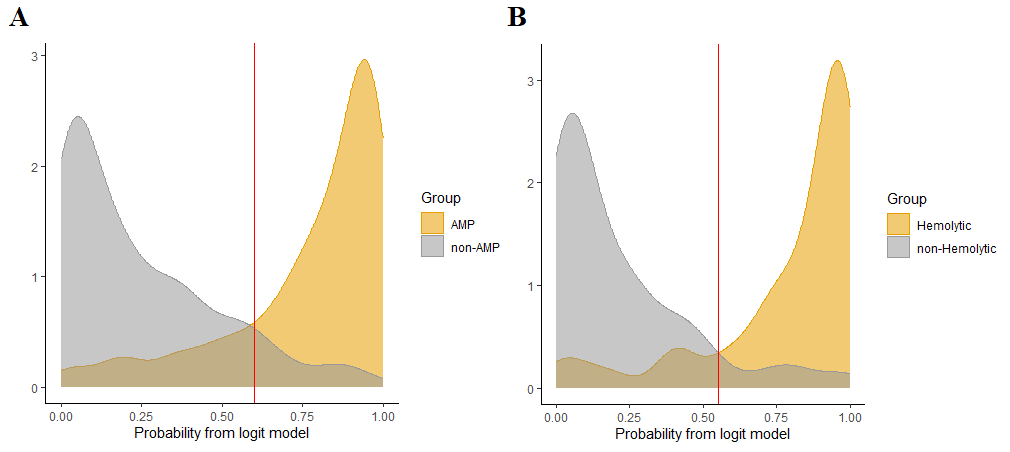
Results of antimicrobial and hemolytic activity prediction (validation on test sets):
A – Performance of the model for antimicrobial activity prediction; B – Performance of the model for hemolytic activity prediction
orange density plots correspond to positive groups and grey plots correspond to negative groups; red vertical lines represent optimal cut-offs for distinguishing between positive and negative groups (0.6 for the AMPs model and 0.55 for the HPs model)
3.2. Results of mature AMPs identification in their precursor proteins
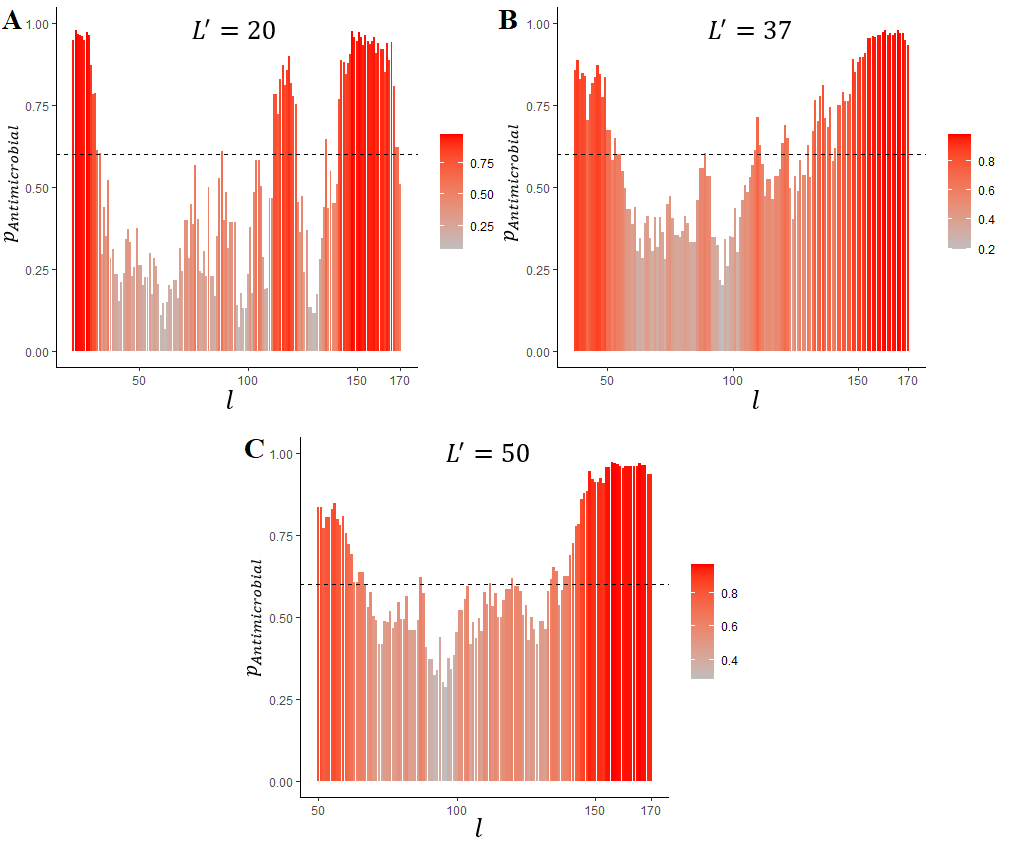
Prediction of LL-37 localization in its precursor protein:
l is the rightmost a.a. in the sliding window, pAntimicrobial is probability of antimicrobial activity, L' is the sliding window length (A – 20, B – 37 and C – 50 amino acid a.a.)
the actual mature LL-37 length is 37 a.a. The dashed horizontal line represents the estimated optimal cut-off for AMPs prediction (0.6)
Prediction of HNP1 localization in its precursor protein:
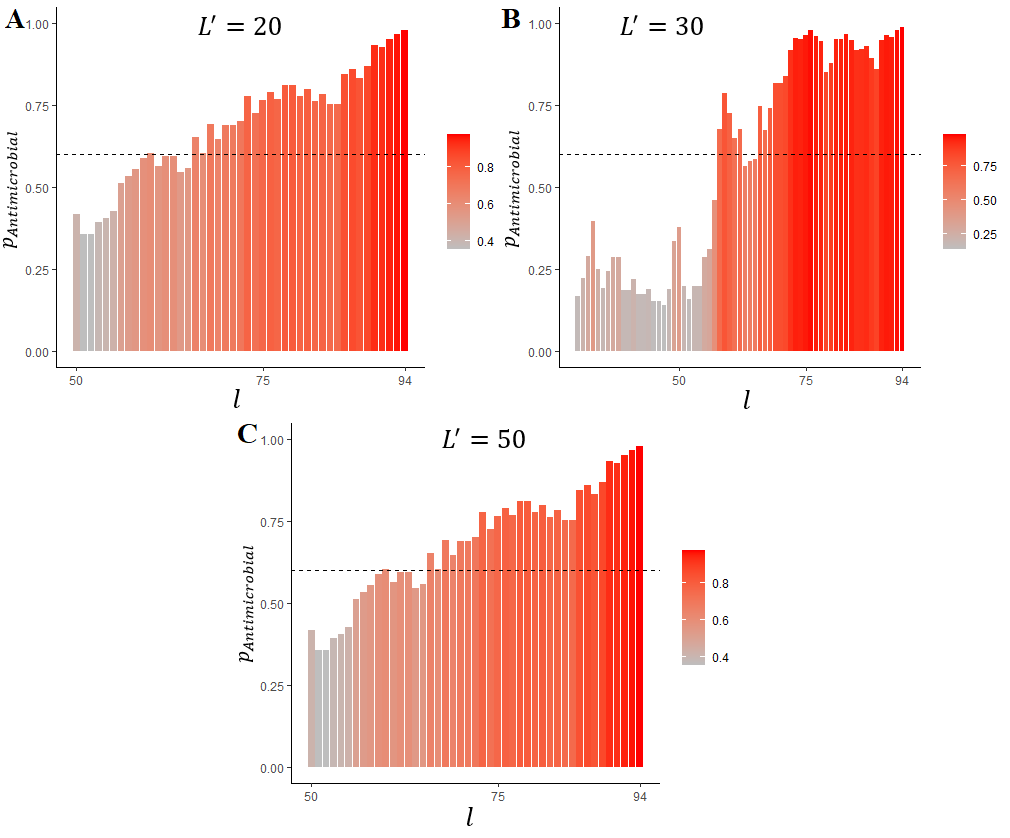
l is the rightmost a.a. in the sliding window, pAntimicrobial is probability of antimicrobial activity, L' is the sliding window length (A – 20 a.a.; B – 30 a.a. and C – 50 a.a.)
the actual mature HNP1 length is 30 a.a.; the dashed horizontal line represents the estimated optimal cut-off for AMPs prediction (0.6)
Prediction of Tachyplesin I localization in its precursor protein:
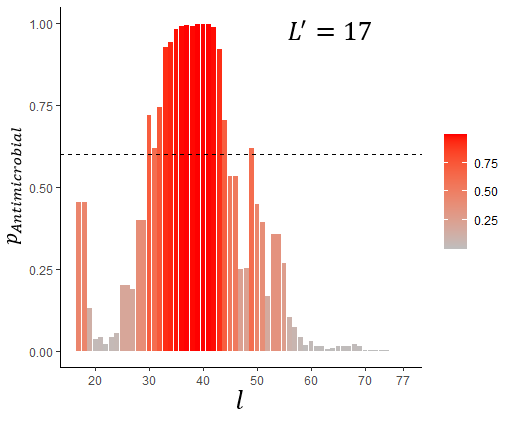
l is the rightmost a.a. in the sliding window, pAntimicrobial is probability of antimicrobial activity, L' is the sliding window length (the only variant corresponding to the actual tacheplesin I length, i.e. 17 a.a.)
the dashed horizontal line represents the estimated optimal cut-off for AMPs prediction (0.6)
4. Discussion
The study includes two successive parts: ML and AMPs prediction from large proteins. The main idea used for prediction of AMPs and HPs was to reveal key structural and physicochemical parameters which are different between positive and negative peptides and to use them as predictors in logistic regression. As expected, predictors in the two models appeared to be co-directed (see Section 3.1). The effects of the selected features are consistent with typical parameters of AMPs widely described in literature (see Introduction) and are in accordance with the distribution of the peptides characteristics illustrated in Fig. 4 and Fig 5. Ideally, prediction of hemolytic activity should be based on direct comparison of hemolytic and non-hemolytic AMPs, which would require a special database.
In the present study, only antibacterial peptides were selected from the original database, but both gram-positive and gram-negative bacteria were included as targets. Indeed, the vast majority of the peptides in the original database were active against these two groups of microorganisms simultaneously. A negligible portion of AMPs were active strictly against one target group other than bacteria (this was the case for fungi, viruses but was not observed for cancers, mammalian cells and parasites). Most predictive tools deal with general antimicrobial activity, although some algorithms attempt to distinguish between AMPs activities by performing multi-label classification , . Nevertheless, this approach was considered as redundant in the present study since one of the main traits of most AMPs is their wide spectrum of activity – antiviral, antiparasitic, antifungal, antitumor, often simultaneously . As such, the built dataset may be relevant for description of AMPs with a wide spectrum of taxonomic targets.
In this study, cytotoxic activity was identified with hemolytic activity, which is not completely correct. Actually, different cell lines and red blood cells from different species demonstrate different susceptibility to the peptides’ membranolytic action depending on their membrane lipid content . However, it is difficult to take into account all possible side targets of AMPs due to their enormous variety, and hemolytic activity is a standard measure of AMPs toxic capacity. It should also be noted that cytotoxicity is not the sole limitation of AMPs implementation into the clinic. Stability in vivo, action on humoral targets and a high cost of synthesis should be controlled during elaboration of novel AMPs.
We intended to elaborate an approach to reveal AMPs in long protein sequences. To validate the proposed model, we used precursor sequences of well-described AMPs because such sequences undoubtedly contain either non-antimicrobial or antimicrobial fragments corresponding to mature AMPs. The lengths of LL-37 and HNPs lie within the range 20–50 used for ML, so the sliding windows lengths applied for the sequences' analysis were 20 (the minimum possible size), 50 (the maximum possible size) and exactly equal to the AMP’s length. In both cases, the C-terminal localization of the AMPs was correctly predicted (Fig. 8–10). Tachiplesin I was used as an example of AMP which is located in the middle but not at the C-terminus of its precursor. This AMP includes 17 a.a. and is shorter than peptides participating in ML. Nevertheless, an excellent prediction was made by the algorithm (Fig. 10). To sum up the results of the prediction, the proposed algorithm is effective at search for AMPs in proteins but many false positive discoveries were detected, especially for LL-37 and HNP1 prediction. Adjustment of the sliding window lengths and the cut-off levels may be beneficial for a particular research task solution. It should be also noted that antimicrobial activity can be possessed not only by the natural AMPs of the given lengths but also by slightly longer or shorter peptides. The preservation of activity of shorter AMPs derived from their longer parents is a desirable strategy but also a common challenge in AMPs elaboration pipelines , .
In principle, the suggested algorithm may be used for a) discovery of natural AMPs primarily serving as defensive molecules of immune systems in different species; b) revealing endogenous peptides with possible antimicrobial function releasing from proteins which are known to undergo proteolytic cleavage, including that related to immune response (complement cascade , blood clotting , etc.); c) search for antimicrobial domains in large proteins with miscellaneous functions. In all cases, the revealed peptides can be used as templates for further design of AMP-based drugs.
5. Conclusion
In this study, we proposed an approach combining ML by multiple LR and AMPs recognition in large proteins, namely in their precursors. Firstly, two predictive models of acceptable quality were constructed. Secondly, the use of the equations coefficients allowed to identify regions corresponding to mature AMPs correctly, although further improvements may be beneficial for enhancement of prediction quality.